In dieser Schritt-für-Schritt-Anleitung erfahren Sie, wie Sie mit Apache Beam, Google Dataflow und TensorFlow ein Modell für maschinelles Lernen vorverarbeiten, trainieren und damit Vorhersagen erstellen.
Zur Veranschaulichung dieser Konzepte wird hier das Codebeispiel "Molecules" verwendet. Mit Daten über Moleküle als Eingabe wird mit dem Codebeispiel "Molecules" ein Modell für maschinelles Lernen zur Vorhersage molekularer Energie erstellt und trainiert.
Kosten
In dieser Schritt-für-Schritt-Anleitung werden möglicherweise kostenpflichtige Komponenten von Google Cloud verwendet, darunter eine oder mehrere der folgenden:
- Dataflow
- Cloud Storage
- AI Platform
Sie können mithilfe des Preisrechners eine Kostenschätzung für Ihre voraussichtliche Nutzung erstellen.
Übersicht
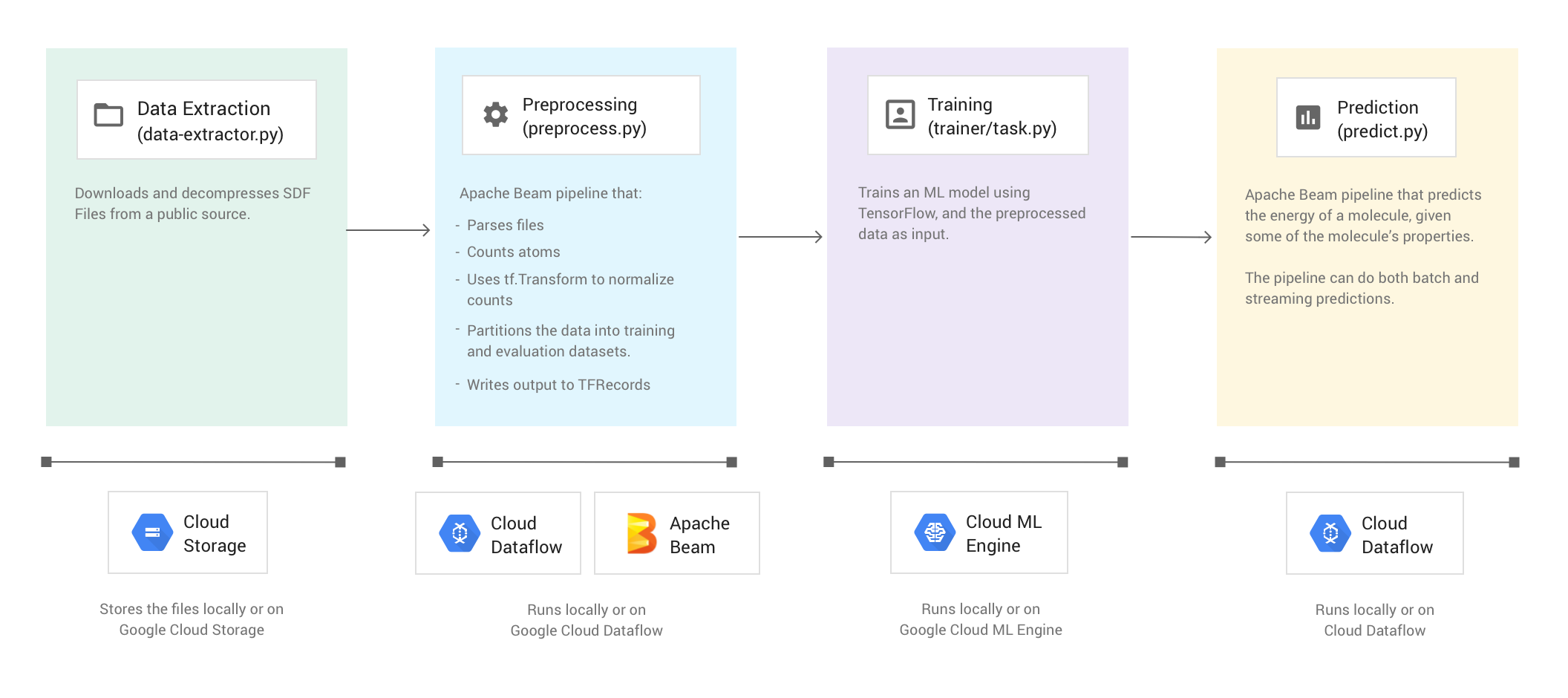
Mit dem Codebeispiel "Molecules" werden Dateien extrahiert, die Daten über Moleküle enthalten, und die jeweilige Anzahl der Kohlenstoff-, Wasserstoff-, Sauerstoff- und Stickstoffatome in jedem Molekül wird gezählt. Dann werden mit dem Code die jeweiligen Anzahlen auf Werte zwischen 0 und 1 normalisiert. Anschließend werden diese Werte in einen TensorFlow Deep Neural Network Estimator eingespeist. Der Neural Network Estimator trainiert ein Modell für maschinelles Lernen zur Vorhersage molekularer Energie.
Das Codebeispiel umfasst vier Phasen:
- Datenextraktion (
data-extractor.py) - Vorverarbeitung (
preprocess.py) - Training (
trainer/task.py) - Vorhersage (
predict.py)
In den folgenden Abschnitten werden alle vier Phasen behandelt. Der Schwerpunkt liegt in dieser Anleitung aber auf den Phasen, in denen Apache Beam und Dataflow zum Einsatz kommen: die Vorverarbeitungsphase und die Vorhersagephase. In der Vorverarbeitungsphase wird auch die TensorFlow Transform-Bibliothek tf.Transform verwendet.
In der folgenden Abbildung wird der Workflow des Codebeispiels "Molecules" veranschaulicht.
Codebeispiel ausführen
Folgen Sie bei der Einrichtung der Umgebung der Anleitung in der README-Datei im GitHub-Repository "Molecules". Führen Sie anschließend das Codebeispiel "Molecules" mit einem der bereitgestellten Wrapper-Skripts run-local oder run-cloud aus. Mit diesen Skripts werden automatisch alle vier Phasen des Codebeispiels ausgeführt (Extraktion, Vorverarbeitung, Training und Vorhersage).
Alternativ können Sie alle Phasen mit den Befehlen in den entsprechenden Abschnitten dieses Dokuments manuell ausführen.
Lokal ausführen
Verwenden Sie das Wrapper-Skript run-local, um das Codebeispiel "Molecules" lokal auszuführen:
./run-local
In den Ausgabelogs wird angezeigt, welche der vier Phasen (Datenextraktion, Vorverarbeitung, Training und Vorhersage) gerade vom Skript ausgeführt wird.
Das Skript data-extractor.py hat ein erforderliches Argument für die Anzahl der Dateien.
Der Einfachheit halber haben das Skript run-local und das Skript run-cloud standardmäßig 5 Dateien für dieses Argument. Jede Datei umfasst 25.000 Moleküle. Die vollständige Ausführung des Codebeispiels dauert ca. 3 bis 7 Minuten. Die Ausführungsdauer hängt von der CPU Ihres Computers ab.
In Google Cloud ausführen
Führen Sie das Wrapper-Skript run-cloud aus, um das Codebeispiel "Molecules" auf Google Cloud auszuführen. Alle Eingabedateien müssen sich in Cloud Storage befinden.
Verweisen Sie mit dem Parameter --work-dir auf den Cloud Storage-Bucket:
./run-cloud --work-dir gs://<your-bucket-name>/cloudml-samples/molecules
Phase 1: Datenextraktion
Quellcode: data-extractor.py
Im ersten Schritt werden die Eingabedaten extrahiert. Die angegebenen SDF-Dateien werden mit der Datei data-extractor.py extrahiert und dekomprimiert. In späteren Schritten des Beispiels werden diese Dateien vorverarbeitet und die Daten zum Trainieren und Evaluieren des Modells für maschinelles Lernen verwendet. Die Datei extrahiert die SDF-Dateien aus der öffentlichen Quelle und speichert sie in einem Unterverzeichnis des angegebenen Arbeitsverzeichnisses. Das Standardarbeitsverzeichnis (--work-dir) ist /tmp/cloudml-samples/molecules.
Extrahierte Dateien speichern
Speichern Sie die extrahierten Datendateien lokal:
python data-extractor.py --max-data-files 5
Oder speichern Sie die extrahierten Datendateien an einem Speicherort in Cloud Storage:
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python data-extractor.py --work-dir $WORK_DIR --max-data-files 5
Phase 2: Vorverarbeitung
Quellcode: preprocess.py
Im Codebeispiel "Molecules" wird eine Apache Beam-Pipeline zur Vorverarbeitung der Daten verwendet. Folgende Vorverarbeitungsaktionen werden mit der Pipeline ausgeführt:
- Lesen und Parsen der extrahierten SDF-Dateien
- Zählen der verschiedenen Atome in jedem der Moleküle in den Dateien
- Normalisieren der Anzahlen mit
tf.Transformauf Werte zwischen 0 und 1 - Partitionieren des Datasets in ein Trainings-Dataset und ein Evaluations-Dataset
- Schreiben der beiden Datasets als Objekte vom Typ
TFRecord
Mit Apache Beam-Transformationen lassen sich einzelne Elemente auf effiziente Weise bearbeiten. Allerdings lassen sich Transformationen, die einen vollständigen Durchlauf des Datasets erfordern, nicht gut mit Apache Beam ausführen. Besser eignet sich dafür tf.Transform. Aus diesem Grund werden im Code Apache Beam-Transformationen zum Lesen und Formatieren der Moleküle sowie zum Zählen der Atome in den einzelnen Molekülen verwendet. Der Code sucht dann mit tf.Transform nach den globalen Mindest- und Höchstwerten, um die Daten zu normalisieren.
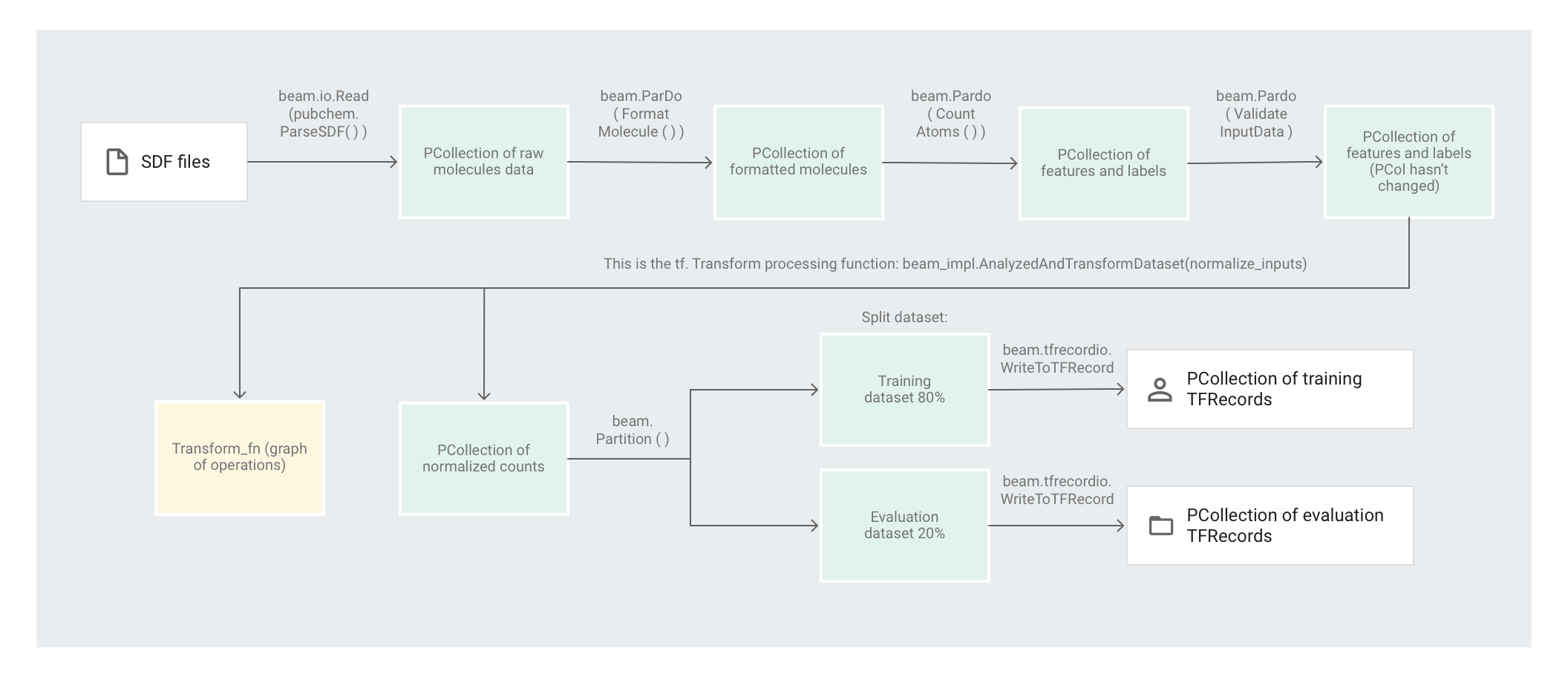
In der folgenden Abbildung sind die Schritte in der Pipeline dargestellt.
Elementbasierte Transformationen anwenden
Mit dem Code preprocess.py wird eine Apache Beam-Pipeline erstellt.
Als Nächstes wird mit dem Code die Transformation feature_extraction auf die Pipeline angewendet.
Für die Transformation feature_extraction nutzt die Pipeline SimpleFeatureExtraction.
Die in pubchem/pipeline.py definierte Transformation SimpleFeatureExtraction umfasst eine Reihe von Transformationen, mit denen alle Elemente voneinander unabhängig bearbeitet werden.
Als Erstes parst der Code die Moleküle aus der Quelldatei, formatiert die Moleküle anschließend zu einem Wörterbuch der Molekülattribute und zählt schließlich die Atome im Molekül. Die jeweilige Anzahl sind die Features (Eingaben) für das Modell für maschinelles Lernen.
Mit der Lesetransformation beam.io.Read(pubchem.ParseSDF(data_files_pattern)) werden SDF-Dateien aus einer benutzerdefinierten Quelle gelesen.
Die benutzerdefinierte Quelle mit dem Namen ParseSDF ist in pubchem/pipeline.py definiert.
ParseSDF dient als Erweiterung von FileBasedSource. Damit wird die Funktion read_records zum Öffnen der extrahierten SDF-Dateien implementiert.
Wenn Sie das Codebeispiel "Molecules" mit Google Cloud ausführen, können die Dateien von mehreren Workern (VMs) gleichzeitig gelesen werden. Damit zwei Worker nicht denselben Inhalt in den Dateien lesen, verwendet jede Datei einen range_tracker.
Die Pipeline gruppiert die Rohdaten in Abschnitte mit relevanten Informationen für die nächsten Schritte. Jeder Abschnitt in der geparsten SDF-Datei wird in einem Wörterbuch gespeichert (siehe pipeline/sdf.py), wobei die Schlüssel die Abschnittsnamen und die Werte die Rohinhalte in den Zeilen des entsprechenden Abschnitts sind.
Mit dem Code wird beam.ParDo(FormatMolecule()) auf die Pipeline angewendet. Mit ParDo wird das DoFn-Objekt mit dem Namen FormatMolecule auf jedes Molekül angewendet. FormatMolecule liefert ein Wörterbuch formatierter Moleküle. Das folgende Snippet ist ein Beispiel für ein Element in der Ausgabe-PCollection:
{
'atoms': [
{
'atom_atom_mapping_number': 0,
'atom_stereo_parity': 0,
'atom_symbol': u'O',
'charge': 0,
'exact_change_flag': 0,
'h0_designator': 0,
'hydrogen_count': 0,
'inversion_retention': 0,
'mass_difference': 0,
'stereo_care_box': 0,
'valence': 0,
'x': -0.0782,
'y': -1.5651,
'z': 1.3894,
},
...
],
'bonds': [
{
'bond_stereo': 0,
'bond_topology': 0,
'bond_type': 1,
'first_atom_number': 1,
'reacting_center_status': 0,
'second_atom_number': 5,
},
...
],
'<PUBCHEM_COMPOUND_CID>': ['3\n'],
...
'<PUBCHEM_MMFF94_ENERGY>': ['19.4085\n'],
...
}
Mit dem Code wird anschließend beam.ParDo(CountAtoms()) auf die Pipeline angewendet. Das DoFn-Objekt CountAtoms summiert die Anzahl der Kohlenstoff-, Wasserstoff-, Stickstoff- und Sauerstoffatome jedes Moleküls. CountAtoms gibt eine PCollection von Features und Labels aus.
Hier ein Beispiel für ein Element in der Ausgabe-PCollection:
{
'ID': 3,
'TotalC': 7,
'TotalH': 8,
'TotalO': 4,
'TotalN': 0,
'Energy': 19.4085,
}
Die Pipeline überprüft anschließend die Eingaben. Das DoFn-Objekt ValidateInputData überprüft, ob jedes Element den in input_schema angegebenen Metadaten entspricht. Mit dieser Prüfung wird sichergestellt, dass die Daten im richtigen Format vorliegen, wenn sie in TensorFlow eingespeist werden.
Transformationen mit vollständigem Durchlauf anwenden
Im Codebeispiel "Molecules" wird ein DNN-Regressor zur Erstellung von Vorhersagen verwendet. Allgemein ist es empfehlenswert, die Eingaben zu normalisieren, bevor sie in das ML-Modell eingespeist werden. Die Pipeline normalisiert mit tf.Transform die jeweiligen Anzahlen der verschiedenen Atome auf Werte zwischen 0 und 1. Weitere Informationen zum Normalisieren von Eingaben finden Sie unter Feature scaling.
Für die Normalisierung der Werte ist ein vollständiger Durchlauf des Datasets erforderlich, bei dem die Minimal- und Maximalwerte aufgezeichnet werden. Der Code untersucht dann mit tf.Transform das gesamte Dataset und wendet Transformationen mit vollständigem Durchlauf an.
Damit Sie tf.Transform verwenden können, muss der Code eine Funktion bereitstellen, die die Logik der Transformation enthält, die am Dataset ausgeführt werden soll. In preprocess.py verwendet der Code die von tf.Transform bereitgestellte Transformation AnalyzeAndTransformDataset. Weitere Informationen zur Verwendung von tf.Transform.
In preprocess.py wird als feature_scaling-Funktion normalize_inputs verwendet, die in pubchem/pipeline.py definiert ist. Die Funktion normalisiert mithilfe der tf.Transform-Funktion scale_to_0_1 die Anzahlen auf Werte zwischen 0 und 1.
Manuelles Normalisieren der Daten ist möglich. Wenn das Dataset allerdings groß ist, geht es mit Dataflow schneller. Mit Dataflow kann die Pipeline nach Bedarf auf mehreren Workern (VMs) ausgeführt werden.
Dataset partitionieren
Als Nächstes partitioniert die Pipeline preprocess.py das Dataset in zwei Datasets. Ca. 80 % der Daten werden als Trainingsdaten und ca. 20 % als Evaluationsdaten zugewiesen.
Ausgabe schreiben
Schließlich werden die beiden Datasets (Training und Evaluation) von der Pipeline preprocess.py mit der Transformation WriteToTFRecord geschrieben.
Vorverarbeitungs-Pipeline ausführen
Führen Sie die Vorverarbeitungs-Pipeline lokal aus:
python preprocess.py
Oder führen Sie die Vorverarbeitungspipeline in Dataflow aus:
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python preprocess.py \
--project $PROJECT \
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--work-dir $WORK_DIR
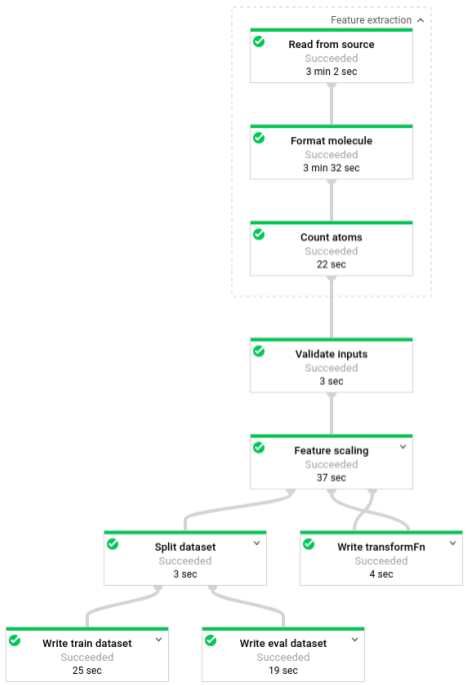
Sobald die Pipeline ausgeführt wird, sehen Sie den Fortschritt der Pipeline in der Dataflow-Monitoring-Oberfläche:
Phase 3: Training
Quellcode: trainer/task.py
Wie bereits erwähnt, erstellt der Code am Ende der Vorverarbeitungsphase zwei Datasets (Training und Evaluation) und zwar durch Aufteilen der Daten.
In dem Beispiel wird TensorFlow zum Trainieren des Modells für maschinelles Lernen verwendet. Die Datei trainer/task.py im Codebeispiel "Molecules" enthält den Code zum Trainieren des Modells. Die Hauptfunktion von trainer/task.py lädt die in der Vorverarbeitungsphase verarbeiteten Daten.
Der Estimator verwendet das Trainings-Dataset zum Trainieren des Modells und anschließend das Evaluations-Dataset, um zu überprüfen, ob das Modell bei bestimmten Molekülattributen die molekulare Energie genau vorhersagt.
Lesen Sie weitere Informationen darüber, wie Sie ein ML-Modell trainieren.
Das Modell trainieren
Trainieren Sie das Modell lokal:
python trainer/task.py
# To get the path of the trained model
EXPORT_DIR=/tmp/cloudml-samples/molecules/model/export/final
MODEL_DIR=$(ls -d -1 $EXPORT_DIR/* | sort -r | head -n 1)
Trainieren Sie das Modell alternativ mit AI Platform:
- Wählen Sie zuerst eine unterstützte Region für die Ausführung des Trainingsjobs aus.
gcloud config set compute/region $REGION
- Trainingsjob starten
JOB="cloudml_samples_molecules_$(date +%Y%m%d_%H%M%S)"
BUCKET=gs://<your bucket name>
WORK_DIR=$BUCKET/cloudml-samples/molecules
gcloud ai-platform jobs submit training $JOB \
--module-name trainer.task \
--package-path trainer \
--staging-bucket $BUCKET \
--runtime-version 1.13 \
--stream-logs \
-- \
--work-dir $WORK_DIR
# To get the path of the trained model:
EXPORT_DIR=$WORK_DIR/model/export
MODEL_DIR=$(gsutil ls -d $EXPORT_DIR/* | sort -r | head -n 1)
Phase 4: Vorhersage
Quellcode: predict.py
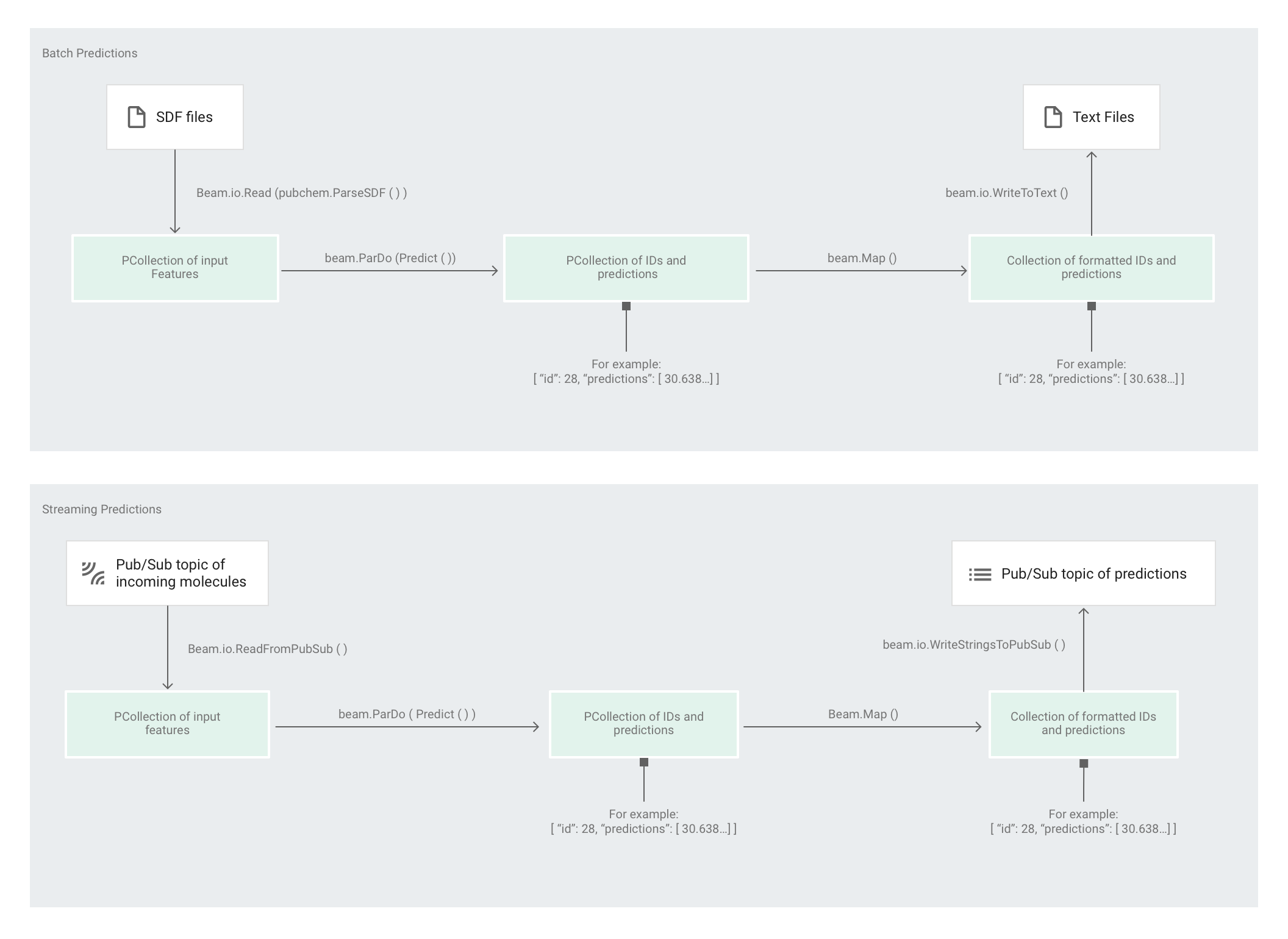
Nachdem das Modell vom Estimator trainiert wurde, können Sie dem Modell Eingaben hinzufügen und Vorhersagen erstellen. Im Codebeispiel "Molecules" werden Vorhersagen von der Pipeline predict.py erstellt. Die Pipeline kann entweder als Batchpipeline oder als Streamingpipeline ausgeführt werden.
Der Code für die Pipeline stimmt für Batch und Streaming zum großen Teil überein, ausgenommen die Abschnitte zu Quell- und Senkeninteraktionen. Wenn die Pipeline im Batchmodus ausgeführt wird, liest sie die Eingabedateien aus der benutzerdefinierten Quelle und schreibt die Ausgabevorhersagen als Textdateien in das angegebene Arbeitsverzeichnis. Wird die Pipeline im Streamingmodus ausgeführt, liest sie die Eingabemoleküle beim Eintreffen aus einem Pub/Sub-Thema. Sobald die Ausgabevorhersagen erstellt sind, schreibt die Pipeline diese in ein anderes Pub/Sub-Thema.
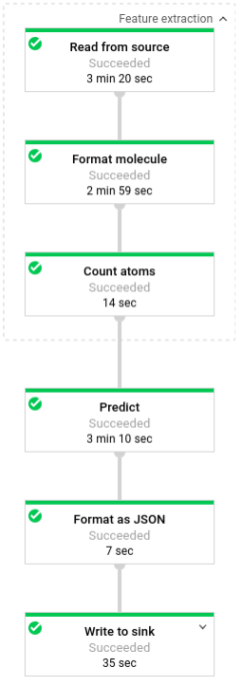
In der folgenden Abbildung sind die Schritte in den Vorhersage-Pipelines (Batch und Streaming) dargestellt.
In predict.py definiert der Code die Pipeline in der Funktion run:
Der Code ruft die Funktion run mit den folgenden Parametern auf:
Zuerst übergibt der Code die Transformation pubchem.SimpleFeatureExtraction(source) als die Transformation feature_extraction. Diese Transformation, die auch in der Vorverarbeitungsphase verwendet wird, wird auf die Pipeline angewandt:
Die Transformation liest basierend auf dem Ausführungsmodus der Pipeline (Batch oder Streaming) die entsprechende Quelle aus, formatiert die Moleküle und zählt die verschiedenen Atome in jedem Molekül.
Anschließend wird beam.ParDo(Predict(…)) auf die Pipeline angewendet, um die molekulare Energie vorherzusagen.
Predict, das übergebene DoFn-Objekt, verwendet das angegebene Wörterbuch der Eingabefeatures (Anzahl von Atomen) zur Vorhersage der molekularen Energie.
Als nächste Transformation wird beam.Map(lambda result: json.dumps(result)) auf die Pipeline angewendet, um das Wörterbuch mit den Vorhersageergebnissen in einen JSON-String zu serialisieren.
Schließlich wird die Ausgabe in die Senke geschrieben (entweder als Textdateien in das Arbeitsverzeichnis für den Batchmodus oder als Nachrichten in ein Pub/Sub-Thema für den Streamingmodus).
Batchvorhersagen
Batchvorhersagen sind für den Durchsatz optimiert, nicht für die Latenz. Am besten funktionieren Batchvorhersagen, wenn Sie viele Vorhersagen erstellen und warten können, bis alle fertig sind, bevor Sie die Ergebnisse erhalten.
Vorhersage-Pipeline im Batchmodus ausführen
Führen Sie die Batchvorhersagepipeline lokal aus:
# For simplicity, we'll use the same files we used for training
python predict.py \
--model-dir $MODEL_DIR \
batch \
--inputs-dir /tmp/cloudml-samples/molecules/data \
--outputs-dir /tmp/cloudml-samples/molecules/predictions
Oder führen Sie die Batchvorhersagepipeline in Dataflow aus:
# For simplicity, we'll use the same files we used for training
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python predict.py \
--work-dir $WORK_DIR \
--model-dir $MODEL_DIR \
batch \
--project $PROJECT \
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--inputs-dir $WORK_DIR/data \
--outputs-dir $WORK_DIR/predictions
Sobald die Pipeline ausgeführt wird, sehen Sie den Fortschritt der Pipeline in der Dataflow-Monitoring-Oberfläche:
Streamingvorhersagen
Streaming-Vorhersagen sind für die Latenz optimiert, nicht für den Durchsatz. Am besten funktionieren Streaming-Vorhersagen, wenn Sie sporadische Vorhersagen erstellen, die Ergebnisse aber so schnell wie möglich erhalten möchten.
Der Vorhersagedienst (die Streaming-Vorhersagepipeline) empfängt die Eingabemoleküle von einem Pub/Sub-Thema und veröffentlicht die Ausgabe (Vorhersagen) in einem anderen Pub/Sub-Thema.
Erstellen Sie das Pub/Sub-Thema für die Eingaben:
gcloud pubsub topics create molecules-inputs
Erstellen Sie das Pub/Sub-Thema für die Ausgaben:
gcloud pubsub topics create molecules-predictions
Führen Sie die Streaming-Vorhersagepipeline lokal aus:
# Run on terminal 1
PROJECT=$(gcloud config get-value project)
python predict.py \
--model-dir $MODEL_DIR \
stream \
--project $PROJECT
--inputs-topic molecules-inputs \
--outputs-topic molecules-predictions
Oder führen Sie die Streaming-Vorhersagepipeline in Dataflow aus:
# Run on terminal 1
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python predict.py \
--work-dir $WORK_DIR \
--model-dir $MODEL_DIR \
stream \
--project $PROJECT
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--inputs-topic molecules-inputs \
--outputs-topic molecules-predictions
Nachdem Sie den Vorhersagedienst (die Streaming-Vorhersagepipeline) gestartet haben, ist es erforderlich, dass Sie einen Publisher ausführen, der Moleküle an den Vorhersagedienst sendet, und einen Subscriber, der die Vorhersageergebnisse abhört. Das Codebeispiel "Molecules" stellt Publisher-Dienste (publisher.py) und Subscriber-Dienste (subscriber.py) zur Verfügung.
Der Publisher parst SDF-Dateien aus einem Verzeichnis und veröffentlicht sie im Eingabethema. Der Subscriber hört die Vorhersageergebnisse ab und protokolliert sie. Zur Vereinfachung werden in diesem Beispiel dieselben SDF-Dateien wie in der Trainingsphase verwendet.
Führen Sie den Subscriber aus:
# Run on terminal 2
python subscriber.py \
--project $PROJECT \
--topic molecules-predictions
Führen Sie den Publisher aus:
# Run on terminal 3
python publisher.py \
--project $PROJECT \
--topic molecules-inputs \
--inputs-dir $WORK_DIR/data
Nachdem der Publisher mit dem Parsen und Veröffentlichen von Molekülen begonnen hat, sehen Sie Vorhersagen des Subscribers.
Bereinigen
Wenn die Ausführung der Streaming-Vorhersagen-Pipeline abgeschlossen ist, stoppen Sie die Pipeline, damit keine Gebühren anfallen.
Weitere Informationen
- Apache Beam-Dokumentation
- TensorFlow-Dokumentation:
- Weiteres Codebeispiel zur Verwendung von Apache Beam und
tf.Transform. - Trainieren Sie ein ML-Modell für die Zeitachsenklassifizierung mit dem Global Fishing Watch-Beispiel.