このチュートリアルでは、Apache Beam、Google Dataflow、TensorFlow を使用して機械学習モデルの前処理、トレーニング、予測を行う方法を示します。
これらのコンセプトを実際に示すために、このチュートリアルでは Molecules コードサンプルを使用します。Molecules コードサンプルは、分子データが入力されると、分子エネルギーを予測する機械学習モデルを作成してトレーニングします。
費用
このチュートリアルでは、Google Cloud の課金対象となる次のコンポーネントを 1 つ以上使用します。
- Dataflow
- Cloud Storage
- AI Platform
料金計算ツールを使うと、予想使用量に基づいて費用の見積もりを出すことができます。
概要
Molecules コードサンプルは、分子データを含むファイルを抽出し、各分子に含まれる炭素、水素、酸素、窒素の原子数をカウントします。その後、コードは、カウントした数を 0 と 1 の間の値に正規化し、その値を TensorFlow Estimator ディープ ニューラル ネットワークに入力します。ニューラル ネットワーク Estimator は、分子エネルギーを予測する目的で機械学習モデルをトレーニングします。
サンプルコードは 4 つのフェーズで構成されています。
- データ抽出(
data-extractor.py) - 前処理(
preprocess.py) - トレーニング(
trainer/task.py) - 予測(
predict.py)
下のセクションでは 4 つのフェーズについて説明しますが、このチュートリアルでは、Apache Beam と Dataflow を使用するフェーズ(前処理フェーズと予測フェーズ)について詳しく説明します。また、前処理フェーズでは TensorFlow Transform ライブラリ(tf.Transform)も使用されます。
次の図は、Molecules コードサンプルのワークフローを示しています。
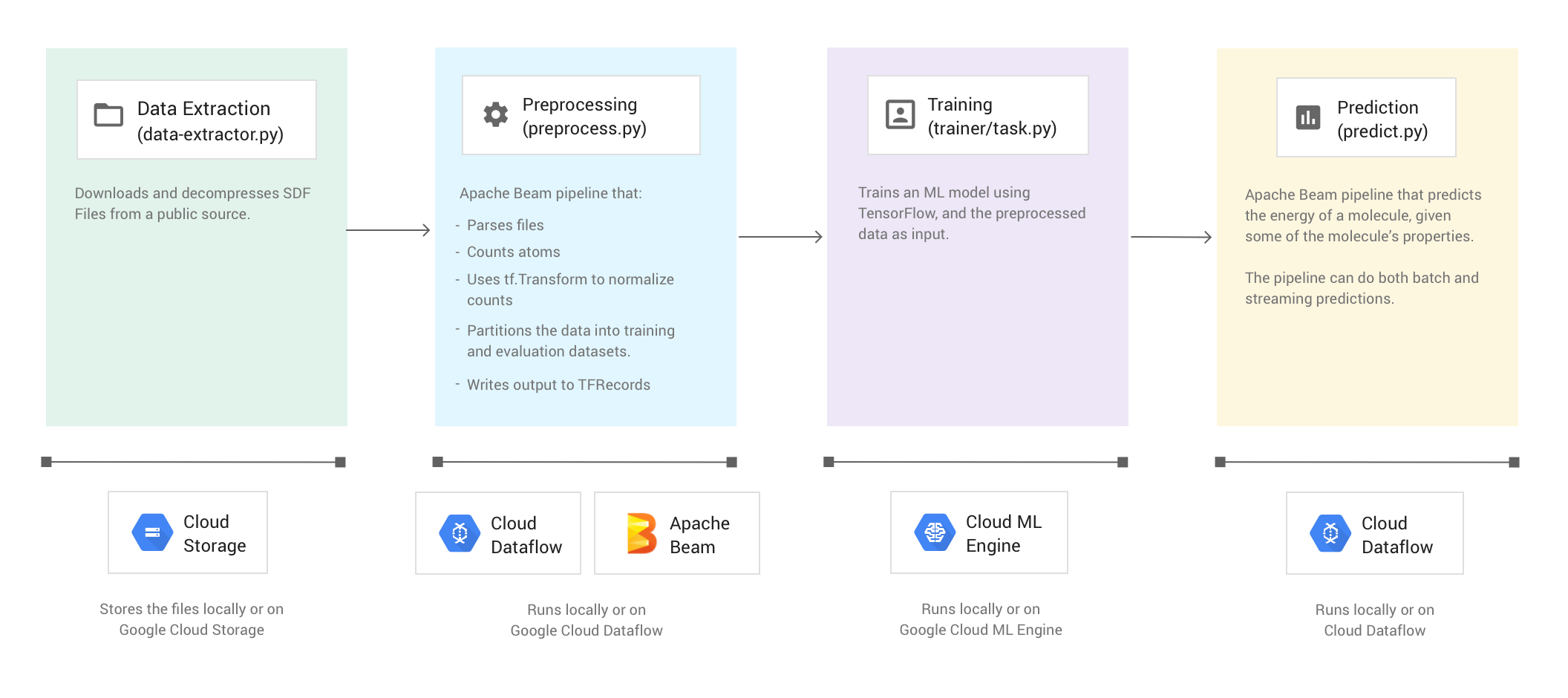
サンプルコードの実行
環境を設定するには、Molecules GitHub リポジトリの README にある指示に従ってください。次に、提供されているラッパー スクリプト run-local または run-cloud を使用して Molecules コードサンプルを実行します。これらのスクリプトは、コードサンプルの 4 つのフェーズ(抽出、前処理、トレーニング、予測)をすべて自動的に実行します。
あるいは、このドキュメントの各セクションに記載されているコマンドを使用して、各フェーズを手動で実行することもできます。
ローカルでの実行
Molecules コードサンプルをローカルで実行するには、run-local ラッパー スクリプトを実行します。
./run-local
スクリプトが 4 つの各フェーズ(データ抽出、前処理、トレーニング、予測)を実行すると、出力ログにそれが表示されます。
data-extractor.py スクリプトには、ファイル数を示す必須の引数があります。run-local スクリプトと run-cloud スクリプトには、簡単に使用できるように、この引数のデフォルト(5 ファイル)が用意されています。各ファイルには 25,000 個の分子が含まれています。コードサンプルを実行すると、開始してから終了するまでに約 3~7 分かかります。実行時間は、お使いのパソコンの CPU によって異なります。
Google Cloud での実行
Google Cloud 上で Molecules コードサンプルを実行するには、run-cloud ラッパー スクリプトを実行します。すべての入力ファイルを Cloud Storage に配置する必要があります。
Cloud Storage バケットに --work-dir パラメータを設定します。
./run-cloud --work-dir gs://<your-bucket-name>/cloudml-samples/molecules
フェーズ 1: データ抽出
ソースコード: data-extractor.py
最初のステップは、入力データの抽出です。data-extractor.py ファイルは、指定された SDF ファイルを抽出して解凍します。後続の手順では、これらのファイルを前処理し、そのデータを使用して機械学習モデルをトレーニングして評価します。このファイルは、パブリック ソースから SDF ファイルを抽出し、指定された作業ディレクトリ内のサブディレクトリにそれらを保存します。デフォルトの作業ディレクトリ(--work-dir)は /tmp/cloudml-samples/molecules です。
抽出されたファイルを格納する
次のように、抽出したデータファイルをローカルに保存します。
python data-extractor.py --max-data-files 5
または、次のように、抽出したデータファイルを Cloud Storage の場所に保存します。
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python data-extractor.py --work-dir $WORK_DIR --max-data-files 5
フェーズ 2: 前処理
ソースコード: preprocess.py
Molecules コードサンプルは、1 つの Apache Beam パイプラインを使用してデータを前処理します。このパイプラインは次の前処理アクションを実行します。
- 抽出された SDF ファイルを読み取って解析します。
- ファイル内の各分子の中で異なる原子の数をカウントします。
tf.Transformを使用して、カウントした数を 0 と 1 の間の値に正規化します。- データセットをトレーニング データセットと評価データセットに分けます。
- 2 つのデータセットを
TFRecordオブジェクトとして書き込みます。
Apache Beam 変換では、一度に 1 つの要素を効率的に処理できますが、データセットの全走査を必要とする変換は Apache Beam だけでは簡単に行えず、tf.Transform を使用する必要があります。そのため、このコードでは、分子の読み取りとフォーマット設定、各分子に含まれる原子のカウントを行うために Apache Beam 変換を使用します。次に、tf.Transform を使用してグローバル最小値と最大値を検索し、データを正規化します。
次の図は、パイプラインのステップを示しています。
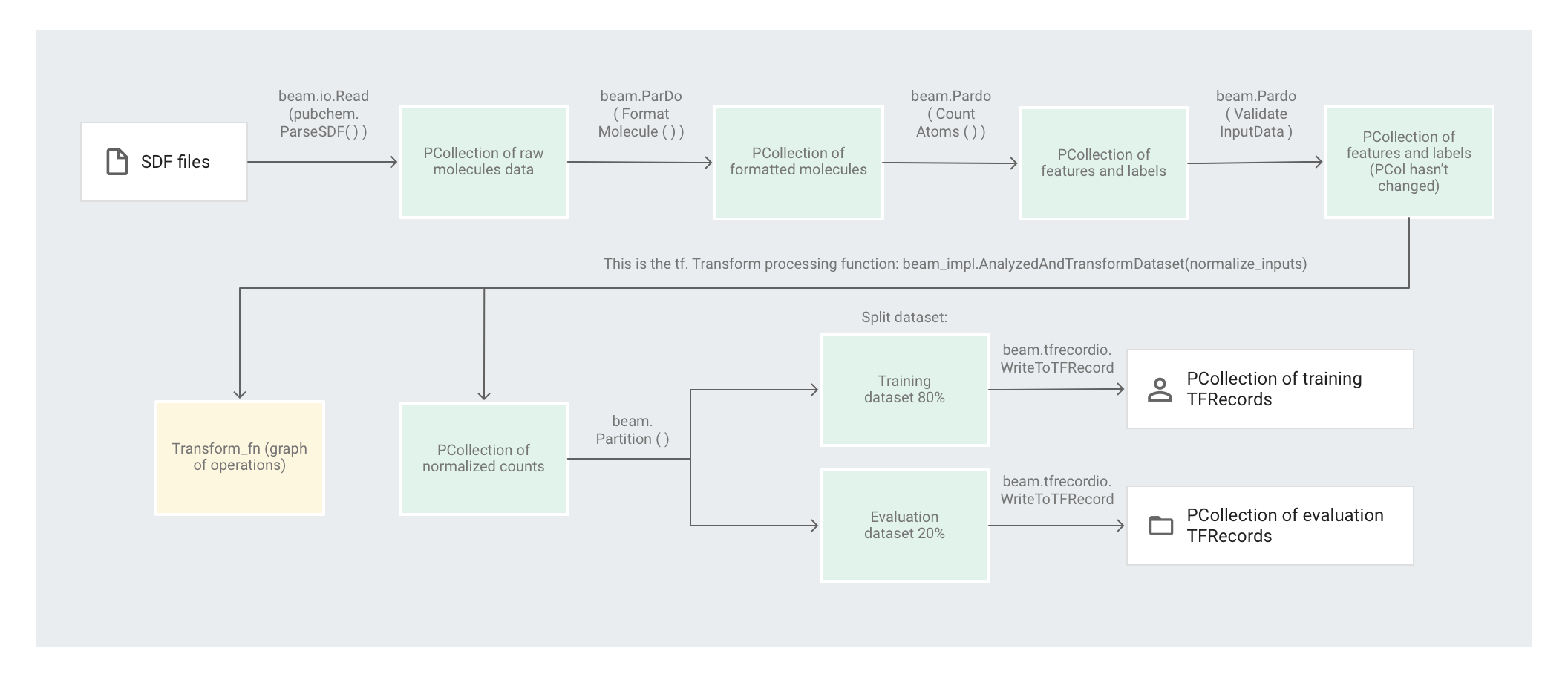
要素ベースの変換の適用
preprocess.py コードは、1 つの Apache Beam パイプラインを作成します。
次に、コードは feature_extraction 変換をパイプラインに適用します。
パイプラインは SimpleFeatureExtraction を feature_extraction 変換として使用します。
pubchem/pipeline.py で定義される SimpleFeatureExtraction 変換には、すべての要素を個別に処理する一連の変換が含まれています。まず、コードはソースファイルの分子を解析して、その分子を分子特性の辞書に合わせてフォーマットし、最後に分子内の原子をカウントします。これらのカウントが機械学習モデルの特徴(入力)になります。
読み取り変換 beam.io.Read(pubchem.ParseSDF(data_files_pattern)) は、カスタムソースから SDF ファイルを読み取ります。
ParseSDF というカスタムソースが pubchem/pipeline.py で定義されています。ParseSDF は FileBasedSource を拡張し、抽出後の SDF ファイルを開く read_records 関数を実装します。
Google Cloud で Molecules コードサンプルを実行する際は、複数のワーカー(VM)が同時にファイルを読み取ることが可能です。2 つのワーカーがファイル内の同じコンテンツを読み取ることがないようにするために、各ファイルは range_tracker を使用します。
パイプラインは、元データを、次のステップで必要になる関連情報のセクションにグループ化します。解析された SDF ファイルの各セクションが辞書に保存されます(pipeline/sdf.py をご覧ください)。ここで、キーはセクション名、値は対応するセクションの元の行の内容です。
このコードは beam.ParDo(FormatMolecule()) をパイプラインに適用します。ParDo は、FormatMolecule という名前の DoFn を各分子に適用します。FormatMolecule は、フォーマットされた分子からなる辞書を生成します。次のスニペットは、出力 PCollection の要素の例です。
{
'atoms': [
{
'atom_atom_mapping_number': 0,
'atom_stereo_parity': 0,
'atom_symbol': u'O',
'charge': 0,
'exact_change_flag': 0,
'h0_designator': 0,
'hydrogen_count': 0,
'inversion_retention': 0,
'mass_difference': 0,
'stereo_care_box': 0,
'valence': 0,
'x': -0.0782,
'y': -1.5651,
'z': 1.3894,
},
...
],
'bonds': [
{
'bond_stereo': 0,
'bond_topology': 0,
'bond_type': 1,
'first_atom_number': 1,
'reacting_center_status': 0,
'second_atom_number': 5,
},
...
],
'<PUBCHEM_COMPOUND_CID>': ['3\n'],
...
'<PUBCHEM_MMFF94_ENERGY>': ['19.4085\n'],
...
}
次に、このコードは beam.ParDo(CountAtoms()) をパイプラインに適用します。DoFn CountAtoms は、各分子が有する炭素、水素、窒素、酸素の原子数を合計します。CountAtoms は、特徴とラベルからなる PCollection を出力します。出力 PCollection の要素の例を次に示します。
{
'ID': 3,
'TotalC': 7,
'TotalH': 8,
'TotalO': 4,
'TotalN': 0,
'Energy': 19.4085,
}
パイプラインは入力を検証します。ValidateInputData DoFn は、input_schema で指定されたメタデータに各要素が一致することを検証します。この検証により、データの形式が TensorFlow に供給される際の正しい形式であることを保証します。
全走査変換の適用
Molecules コードサンプルは、ディープ ニューラル ネットワーク リグレッサーを使用して予測を行います。通常、ML モデルに供給する前に入力を正規化することが推奨されています。パイプラインは tf.Transform を使用して、各原子のカウントを 0 と 1 の間の値に正規化します。入力の正規化の詳細については、特徴スケーリングをご覧ください。
値を正規化するには、最小値と最大値を記録するデータセットを介した全走査が必要になります。コードは tf.Transform を使用して、データセット全体を通過し、全走査変換を適用します。
tf.Transform を使用するには、データセットに対して実行する変換のロジックを含む関数がコードで提供されている必要があります。preprocess.py では、tf.Transform によって提供される AnalyzeAndTransformDataset 変換を使用します。tf.Transform の使用方法をご覧ください。
preprocess.py で、使用される feature_scaling 関数は normalize_inputs で、これは pubchem/pipeline.py に定義されています。この関数は、tf.Transform 関数 scale_to_0_1を使用して、カウントを 0 から 1 の間の値に正規化します。
手動でデータを正規化することは可能ですが、データセットが大きい場合は、Dataflow を使用したほうが速く処理できます。Dataflow を使用すると、必要に応じて複数のワーカー(VM)上でパイプラインを実行できます。
データセットのパーティショニング
次に、preprocess.py パイプラインは、1 つのデータセットを 2 つのデータセットに分けます。トレーニング データとして使われるデータに約 80%、評価データとして使われるデータに約 20% を割り当てます。
出力の書き込み
最後に、preprocess.py パイプラインは、WriteToTFRecord 変換を使用して 2 つのデータセット(トレーニングと評価)を書き込みます。
前処理パイプラインの実行
次のように、ローカルで前処理パイプラインを実行します。
python preprocess.py
あるいは、次のように Dataflow で前処理パイプラインを実行します。
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python preprocess.py \
--project $PROJECT \
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--work-dir $WORK_DIR
パイプラインの実行後、Dataflow モニタリング インターフェースでパイプラインの進行状況を確認できます。
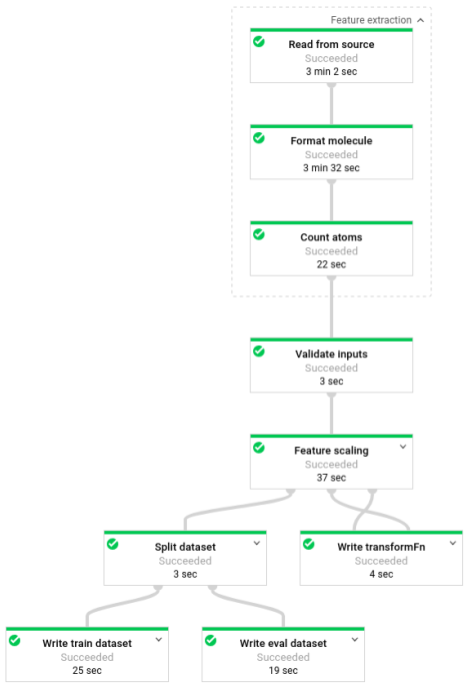
フェーズ 3: トレーニング
ソースコード: trainer/task.py
前処理フェーズの終わりに、コードで 2 つのデータセット(トレーニングと評価)にデータを分割したことに注意してください。
このサンプルでは、TensorFlow を使用して機械学習モデルをトレーニングします。Molecules コードサンプル内の trainer/task.py ファイルには、モデルをトレーニングするためのコードが含まれています。trainer/task.py のメイン関数は、前処理フェーズで処理されたデータを読み込みます。
Estimator は、まずトレーニング データセットを使ってモデルをトレーニングします。次に評価データセットを使用して、いくつかの分子特性に基づきモデルが分子エネルギーを正確に予測するかどうか検証します。
ML モデルのトレーニングで詳細をご覧ください。
モデルのトレーニング
次のように、ローカルでモデルをトレーニングします。
python trainer/task.py
# To get the path of the trained model
EXPORT_DIR=/tmp/cloudml-samples/molecules/model/export/final
MODEL_DIR=$(ls -d -1 $EXPORT_DIR/* | sort -r | head -n 1)
あるいは、次のように AI Platform でモデルをトレーニングします。
- まず、トレーニング ジョブを実行するサポート対象リージョンを選択します
gcloud config set compute/region $REGION
- トレーニング ジョブを起動します
JOB="cloudml_samples_molecules_$(date +%Y%m%d_%H%M%S)"
BUCKET=gs://<your bucket name>
WORK_DIR=$BUCKET/cloudml-samples/molecules
gcloud ai-platform jobs submit training $JOB \
--module-name trainer.task \
--package-path trainer \
--staging-bucket $BUCKET \
--runtime-version 1.13 \
--stream-logs \
-- \
--work-dir $WORK_DIR
# To get the path of the trained model:
EXPORT_DIR=$WORK_DIR/model/export
MODEL_DIR=$(gsutil ls -d $EXPORT_DIR/* | sort -r | head -n 1)
フェーズ 4: 予測
ソースコード: predict.py
Estimator がモデルをトレーニングした後、モデルに入力を提供すると、予測が行われます。Molecules コードサンプルでは、predict.py の中のパイプラインが予測を行います。このパイプラインは、バッチ パイプラインまたはストリーミング パイプラインのいずれかとして機能できます。
パイプラインのコードはバッチとストリーミングで同じです(ただしソースとシンクのインタラクションを除く)。パイプラインをバッチモードで実行した場合、パイプラインはカスタムソースから入力ファイルを読み取り、指定された作業ディレクトリに出力予測をテキスト ファイルとして書き込みます。パイプラインをストリーミング モードで実行した場合、入力分子が到着したときに、それを Pub/Sub トピックから読み取ります。パイプラインは、出力予測の準備ができると、それを別の Pub/Sub トピックに書き込みます。
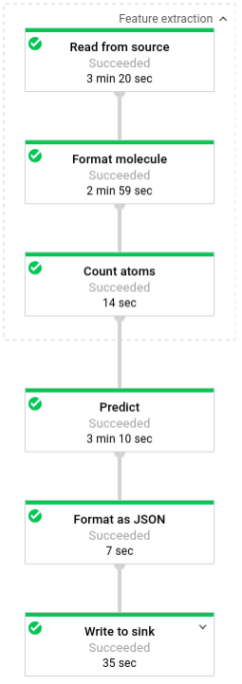
次の図は、予測パイプラインのステップを示しています(バッチおよびストリーミング)。
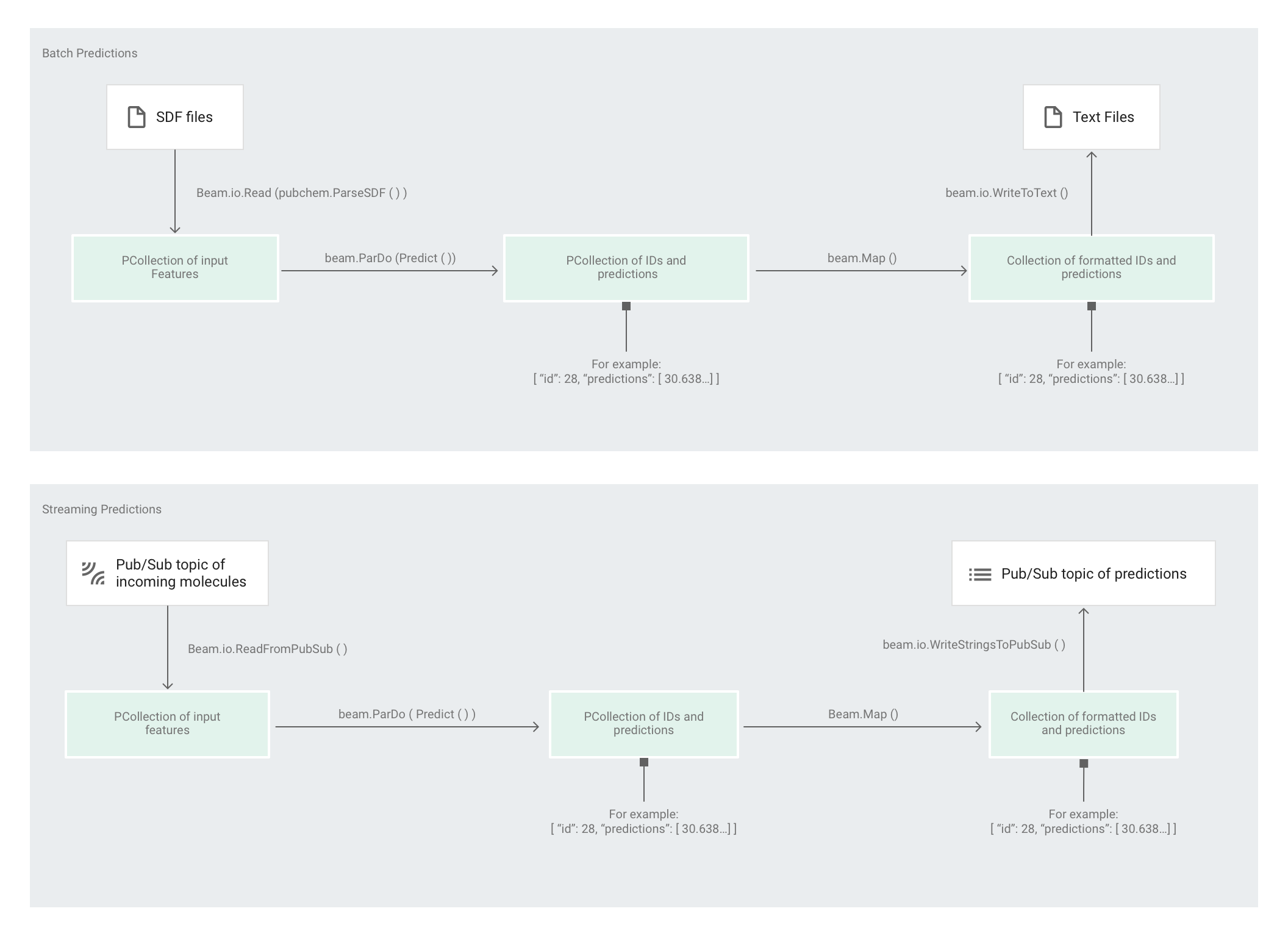
predict.py では、コードの run 関数内でパイプラインが次のように定義されます。
次のパラメータを使用して run 関数が呼び出されます。
まず、このコードは pubchem.SimpleFeatureExtraction(source) 変換を feature_extraction 変換として渡します。前処理フェーズでも使用されたこの変換は、次のようにパイプラインに適用されます。
この変換では、パイプラインの実行モード(バッチまたはストリーミング)に応じて該当するソースから読み取り、分子をフォーマットして、各分子の中の異なる原子をカウントします。
次に beam.ParDo(Predict(…)) が、分子エネルギーの予測を行うパイプラインに適用されます。Predict(渡される DoFn)は、入力特徴(原子のカウント数)からなる特定の辞書を使用して分子エネルギーを予測します。
その次にパイプラインに適用される変換は beam.Map(lambda result: json.dumps(result)) です。これは、予測結果の辞書を受け入れて、それを JSON 文字列にシリアル化します。
最後に、出力がシンクに書き込まれます(バッチモードの場合はテキスト ファイルとして作業ディレクトリに、ストリーミング モードの場合は Pub/Sub トピックに公開されるメッセージとして)。
バッチ予測
バッチ予測は、レイテンシではなくスループットに関して最適化されています。多数の予測を行う場合、すべての予測が完了して結果が得られるまで待つことができるなら、バッチ予測が適しています。
バッチモードでの予測パイプラインの実行
次のように、バッチ予測パイプラインをローカルで実行します。
# For simplicity, we'll use the same files we used for training
python predict.py \
--model-dir $MODEL_DIR \
batch \
--inputs-dir /tmp/cloudml-samples/molecules/data \
--outputs-dir /tmp/cloudml-samples/molecules/predictions
または、次のように Dataflow でバッチ予測パイプラインを実行します。
# For simplicity, we'll use the same files we used for training
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python predict.py \
--work-dir $WORK_DIR \
--model-dir $MODEL_DIR \
batch \
--project $PROJECT \
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--inputs-dir $WORK_DIR/data \
--outputs-dir $WORK_DIR/predictions
パイプラインの実行後、Dataflow モニタリング インターフェースでパイプラインの進行状況を確認できます。
ストリーミング予測
ストリーミング予測は、スループットではなくレイテンシに対して最適化されています。散発的に予測を行い、できるだけ早く結果を得たい場合には、ストリーミング予測が効果的です。
予測サービス(ストリーミング予測パイプライン)は、Pub/Sub トピックから入力分子を受け取り、その出力(予測)を別の Pub/Sub トピックに公開します。
次のように、入力 Pub/Sub トピックを作成します。
gcloud pubsub topics create molecules-inputs
次のように、出力 Pub/Sub トピックを作成します。
gcloud pubsub topics create molecules-predictions
次のように、ストリーミング予測パイプラインをローカルで実行します。
# Run on terminal 1
PROJECT=$(gcloud config get-value project)
python predict.py \
--model-dir $MODEL_DIR \
stream \
--project $PROJECT
--inputs-topic molecules-inputs \
--outputs-topic molecules-predictions
または、次のように Dataflow でストリーミング予測パイプラインを実行します。
# Run on terminal 1
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python predict.py \
--work-dir $WORK_DIR \
--model-dir $MODEL_DIR \
stream \
--project $PROJECT
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--inputs-topic molecules-inputs \
--outputs-topic molecules-predictions
予測サービス(ストリーミング予測パイプライン)を実行した後、分子を予測サービスに送るためにパブリッシャーを実行し、予測結果をリッスンするためにサブスクライバーを実行する必要があります。Molecules コードサンプルには、パブリッシャー(publisher.py)とサブスクライバー(subscriber.py)サービスがあります。
このパブリッシャーは SDF ファイルをディレクトリから解析し、それを入力トピックに公開します。サブスクライバーは予測結果をリッスンし、それらを記録します。わかりやすくするために、この例では、トレーニング フェーズで使用したのと同じ SDF ファイルを使用します。
次のように、サブスクライバーを実行します。
# Run on terminal 2
python subscriber.py \
--project $PROJECT \
--topic molecules-predictions
次のように、パブリッシャーを実行します。
# Run on terminal 3
python publisher.py \
--project $PROJECT \
--topic molecules-inputs \
--inputs-dir $WORK_DIR/data
パブリッシャーが分子の解析と公開を開始すると、サブスクライバーからの予測が表示され始めます。
クリーンアップ
ストリーミング予測パイプラインの実行が完了したら、パイプラインを停止して、料金が発生しないようにします。
次のステップ
- Apache Beam のドキュメントを確認する
- TensorFlow の次のドキュメントを読む
- Apache Beam と
tf.Transformを使用する別のコードサンプルを試す。 - Global Fishing Watch の例を使用して、時系列分類の ML モデルをトレーニングする。