This page explains how to run Sentieon® DNASeq® as a Google Cloud pipeline for secondary genomic analysis. The pipeline matches the following results from the Genome Analysis Toolkit (GATK) Best Practices version 3.7:
- Alignment
- Sorting
- Duplicate removal
- Base quality score recalibration (BQSR)
- Variant calling
Input formats include the following:
- fastq files
- Aligned and sorted BAM files
Objectives
After completing this tutorial, you'll know how to:
- Run a pipeline on Google Cloud using Sentieon® DNASeq®
- Write configuration files for different Sentieon® DNASeq® use cases
Costs
In this document, you use the following billable components of Google Cloud:
- Compute Engine
- Cloud Storage
To generate a cost estimate based on your projected usage,
use the pricing calculator.
Before you begin
- Install Python 2.7+. For more information on setting up your Python development environment, such as installing pip on your system, see the Python Development Environment Setup Guide.
- Sign in to your Google Cloud account. If you're new to Google Cloud, create an account to evaluate how our products perform in real-world scenarios. New customers also get $300 in free credits to run, test, and deploy workloads.
-
In the Google Cloud console, on the project selector page, select or create a Google Cloud project.
-
Make sure that billing is enabled for your Google Cloud project.
-
Enable the Cloud Life Sciences, Compute Engine, and Cloud Storage APIs.
-
In the Google Cloud console, on the project selector page, select or create a Google Cloud project.
-
Make sure that billing is enabled for your Google Cloud project.
-
Enable the Cloud Life Sciences, Compute Engine, and Cloud Storage APIs.
- Install the Google Cloud CLI.
-
To initialize the gcloud CLI, run the following command:
gcloud init -
Update and install
gcloudcomponents:gcloud components update
gcloud components install beta - Install git to download the required files.
-
By default, Compute Engine has resource quotas
in place to prevent inadvertent usage. By increasing quotas, you can launch
more virtual machines concurrently, increasing throughput and reducing
turnaround time.
For best results in this tutorial, you should request additional quota above your project's default. Recommendations for quota increases are provided in the following list alongside the minimum quotas needed to run the tutorial. Make your quota requests in the
us-central1region:- CPUs: 64
- Persistent Disk Standard (GB): 375
You can leave other quota request fields empty to keep your current quotas.
Sentieon® evaluation license
When using this pipeline, Sentieon® automatically grants you a free two week
evaluation license of
its software for use with Google Cloud. To receive the license, enter your
email address in the EMAIL field when configuring the pipeline. See
Understanding the input format for
information on setting this field.
To continue using Sentieon® after the evaluation license expires, contact support@sentieon.com.
Set up your local environment and install prerequisites
If you don't have virtualenv, run the following command to install it using pip:
pip install virtualenv
Run the following command to create an isolated Python environment and install dependencies:
virtualenv env source env/bin/activate pip install --upgrade \ pyyaml \ google-api-python-client \ google-auth \ google-cloud-storage \ google-auth-httplib2
Download the pipeline script
Run the following command to download the example files and set your current directory:
git clone https://github.com/sentieon/sentieon-google-genomics.git cd sentieon-google-genomics
Understand the input format
The pipeline uses parameters specified in a JSON file as its input.
In the repository you downloaded, there is an examples/example.json file
with the following content:
{ "FQ1": "gs://sentieon-test/pipeline_test/inputs/test1_1.fastq.gz", "FQ2": "gs://sentieon-test/pipeline_test/inputs/test1_2.fastq.gz", "REF": "gs://sentieon-test/pipeline_test/reference/hs37d5.fa", "OUTPUT_BUCKET": "gs://BUCKET", "ZONES": "us-central1-a,us-central1-b,us-central1-c,us-central1-f", "PROJECT_ID": "PROJECT_ID" "REQUESTER_PROJECT": "PROJECT_ID", "EMAIL": "YOUR_EMAIL_HERE" }
The following table describes the JSON keys in the file:
| JSON key | Description |
|---|---|
FQ1 |
The first pair of reads in the input fastq file. |
FQ2 |
The second pair of reads in the input fastq file. |
BAM |
The input BAM file, if applicable. |
REF |
The reference genome. If set, the fastq/BAM index files are assumed to exist. |
OUTPUT_BUCKET |
The bucket and directory used to store the data output from the pipeline. |
ZONES |
A comma-separated list of Google Cloud zones to use for the worker node. |
PROJECT_ID |
Your Google Cloud project ID. |
REQUESTER_PROJECT |
A project to bill when transferring data from Requester Pays buckets. |
EMAIL |
Your email address. |
Run the pipeline
In the
sentieon-google-genomicsdirectory, edit theexamples/example.jsonfile, substituting the BUCKET, REQUESTER_PROJECT, EMAIL, and PROJECT_ID variables with the relevant resources from your Google Cloud project:{ "FQ1": "gs://sentieon-test/pipeline_test/inputs/test1_1.fastq.gz", "FQ2": "gs://sentieon-test/pipeline_test/inputs/test1_2.fastq.gz", "REF": "gs://sentieon-test/pipeline_test/reference/hs37d5.fa", "OUTPUT_BUCKET": "gs://BUCKET", "ZONES": "us-central1-a,us-central1-b,us-central1-c,us-central1-f", "PROJECT_ID": "PROJECT_ID", "REQUESTER_PROJECT": "PROJECT_ID", "EMAIL": "EMAIL_ADDRESS" }
Set the PROJECT_ID variable in your environment:
export PROJECT_ID=PROJECT_ID
Run the following command to execute the DNASeq® pipeline on a small test dataset identified by the inputs in the configuration file. By default, the script verifies that the input files exist in your Cloud Storage bucket before starting the pipeline.
python runner/sentieon_runner.py --requester_project $PROJECT_ID examples/example.json
If you specified multiple preemptible tries, the pipeline restarts whenever its instances are preempted. After the pipeline finishes, it outputs a message to the console that states whether the pipeline succeeded or failed.
Recommended configuration
For most situations, you can optimize turnaround time and cost using the following configuration. The configuration runs a 30x human genome at a cost of roughly $1.25 and takes about 2 hours. A human whole exome costs roughly $0.35 and take about 45 minutes. Both of these estimates are based on the pipeline's instances not being preempted.
{ "FQ1": "gs://my-bucket/sample1_1.fastq.gz", "FQ2": "gs://my-bucket/sample1_2.fastq.gz", "REF": "gs://sentieon-test/pipeline_test/reference/hs37d5.fa", "OUTPUT_BUCKET": "gs://BUCKET", "BQSR_SITES": "gs://sentieon-test/pipeline_test/reference/Mills_and_1000G_gold_standard.indels.b37.vcf.gz,gs://sentieon-test/pipeline_test/reference/1000G_phase1.indels.b37.vcf.gz,gs://sentieon-test/pipeline_test/reference/dbsnp_138.b37.vcf.gz", "DBSNP": "gs://sentieon-test/pipeline_test/reference/dbsnp_138.b37.vcf.gz", "PREEMPTIBLE_TRIES": "2", "NONPREEMPTIBLE_TRY": true, "STREAM_INPUT": "True", "ZONES": "us-central1-a,us-central1-b,us-central1-c,us-central1-f", "PROJECT_ID": "PROJECT_ID", "EMAIL": "EMAIL_ADDRESS" }
Additional options
You can customize a pipeline using the following additional options.
Input file options
The pipeline supports multiple comma-separated fastq files as input, as the following configuration shows:
"FQ1": "gs://my-bucket/s1_prep1_1.fastq.gz,gs://my-bucket/s1_prep2_1.fastq.gz",
"FQ2": "gs://my-bucket/s1_prep1_2.fastq.gz,gs://my-bucket/s1_prep2_2.fastq.gz",
The pipeline accepts comma-separated BAM files as an input using the BAM JSON
key. Reads in the BAM files are not aligned to the reference genome.
Instead, they start at the data deduplication stage of the pipeline. The following
sample shows a configuration using two BAM files as an input:
"BAM": "gs://my-bucket/s1_prep1.bam,gs://my-bucket/s1_prep2.bam"
Whole-exome data or large datasets configuration
The settings in the recommended configuration are optimized for human whole-genome samples sequenced to an average coverage of 30x. For files that are much smaller or larger than standard whole-genome datasets, you can increase or decrease the resources available to the instance. For best results with large datasets, use the following settings:
{ "FQ1": "gs://sentieon-test/pipeline_test/inputs/test1_1.fastq.gz", "FQ2": "gs://sentieon-test/pipeline_test/inputs/test1_2.fastq.gz", "REF": "gs://sentieon-test/pipeline_test/reference/hs37d5.fa", "OUTPUT_BUCKET": "gs://BUCKET", "ZONES": "us-central1-a,us-central1-b,us-central1-c,us-central1-f", "PROJECT_ID": "PROJECT_ID", "EMAIL": "EMAIL_ADDRESS", "DISK_SIZE": 600, "MACHINE_TYPE": "n1-highcpu-64", "CPU_PLATFORM": "Intel Broadwell" }
The following table provides a description of the settings used:
| JSON key | Description |
|---|---|
DISK_SIZE |
SSD space available to the worker node. |
MACHINE_TYPE |
The type of Compute Engine virtual machine to use. Defaults to n1-standard-1. |
CPU_PLATFORM |
The CPU platform to request. Must be a valid Compute Engine CPU platform name (such as "Intel Skylake"). |
Preemptible instances
You can use preemptible instances in your pipeline
by setting the PREEMPTIBLE_TRIES JSON key.
By default, the runner tries to run the pipeline with a standard instance
if the preemptible tries are exhausted or if the NONPREEMPTIBLE_TRY JSON key
is set to 0. You can turn off this behavior by setting the
NONPREEMPTIBLE_TRY key to false, as shown in the following configuration:
"PREEMPTIBLE_TRIES": 2,
"NONPREEMPTIBLE_TRY": false
The following table provides a description of the settings used:
| JSON key | Description |
|---|---|
PREEMPTIBLE_TRIES |
The number of times to attempt the pipeline when using preemptible instances. |
NONPREEMPTIBLE_TRY |
Determines whether to try running the pipeline with a standard instance after preemptible tries are exhausted. |
Read groups
Read groups are added when fastq files are aligned with a reference
genome using Sentieon® BWA. You can supply multiple comma-separated read groups.
The number of read groups must match the number of input fastq files.
The default read group is @RG\\tID:read-group\\tSM:sample-name\\tPL:ILLUMINA.
To change the read group, set the READGROUP key in the JSON input file, as
shown in the following configuration:
"READGROUP": "@RG\\tID:my-rgid-1\\tSM:my-sm\\tPL:ILLUMINA,@RG\\tID:my-rgid-2\\tSM:my-sm\\tPL:ILLUMINA"
The following table provides a description of the setting used:
| JSON key | Description |
|---|---|
READGROUP |
A read group containing sample metadata. |
For more information on read groups, see Read groups.
Streaming input from Cloud Storage
You can stream input fastq files from Cloud Storage which can reduce
the total runtime of the pipeline. To stream input fastq files from
Cloud Storage, set the STREAM_INPUT JSON key to True:
"STREAM_INPUT": "True"
The following table provides a description of the setting used:
| JSON key | Description |
|---|---|
STREAM_INPUT |
Determines whether to stream input fastq files directly from Cloud Storage. |
Duplicate marking
By default, the pipeline removes duplicate reads from BAM files. You can
change this behavior by setting the DEDUP JSON key, as shown in the following
configuration:
"DEDUP": "markdup"
The following table provides a description of the setting used:
| JSON key | Description |
|---|---|
DEDUP |
Duplicate marking behavior. Valid values:
|
Base quality score recalibration (BQSR) and known sites
BSQR requires known sites of genetic variation. The default behavior is to
skip this stage of the pipeline. However, you can enable BSQR by supplying
known sites with the BQSR_SITES JSON key. If supplied, a
DBSNP file can be used to annotate the output variants during variant
calling.
"BQSR_SITES": "gs://my-bucket/reference/Mills_and_1000G_gold_standard.indels.b37.vcf.gz,gs://my-bucket/reference/1000G_phase1.indels.b37.vcf.gz,gs://my-bucket/reference/dbsnp_138.b37.vcf.gz",
"DBSNP": "gs://sentieon-test/pipeline_test/reference/dbsnp_138.b37.vcf.gz"
The following table provides a description of the settings used:
| JSON key | Description |
|---|---|
BSQR_SITES |
Turns on BSQR and uses a comma-separated list of supplied files as known sites. |
DBSNP |
A dbSNP file used during variant calling. |
Intervals
For some applications, such as targeted or whole-exome sequencing, you might
be interested only in a portion of the genome. In those cases, supplying a file
of target intervals can speed up processing and reduce low quality off-target
variant calls. You can use intervals with the INTERVAL_FILE and INTERVAL
JSON keys.
"INTERVAL_FILE": "gs://my-bucket/capture-targets.bed",
"INTERVAL": "9:80331190-80646365"
The following table provides a description of the settings used:
| JSON key | Description |
|---|---|
INTERVAL_FILE |
A file containing genomic intervals to process. |
INTERVAL |
A string containing a genomic interval to process. |
Output options
By default, the pipeline produces a preprocessed BAM, quality control
metrics, and variant calls. You can disable any of these outputs using the
NO_BAM_OUTPUT, NO_METRICS, and NO_HAPLOTYPER JSON keys. If the
NO_HAPLOTYPER argument is not supplied or NULL, you can use the
GVCF_OUTPUT JSON key to produce variant calls in gVCF format rather than VCF
format.
"NO_BAM_OUTPUT": "true",
"NO_METRICS": "true",
"NO_HAPLOTYPER": "true",
"GVCF_OUTPUT": "true",
The following table provides a description of the settings used:
| JSON key | Description |
|---|---|
NO_BAM_OUTPUT |
Determines whether to output a preprocessed BAM file. |
NO_METRICS |
Determines whether to output file metrics. |
NO_HAPLOTYPER |
Determines whether to output variant calls. |
GVCF_OUTPUT |
Determines whether to output variant calls in gVCF format. |
Sentieon® DNASeq® versions
You can use any recent version of the Sentieon® DNASeq® software package with the
Cloud Life Sciences API by specifying the SENTIEON_VERSION JSON key, like
so:
"SENTIEON_VERSION": "201808.08"
The following versions are valid:
201711.01201711.02201711.03201711.04201711.05201808201808.01201808.03201808.05201808.06201808.07201808.08
Clean up
After finishing the tutorial, you can clean up the resources you created on Google Cloud so you won't be billed for them in the future. The following sections describe how to delete or turn off these resources.
Delete the project
The easiest way to eliminate billing is to delete the project you used for the tutorial.
To delete the project:
- In the Google Cloud console, go to the Projects page.
-
In the project list, select the project you
want to delete and click Delete project.
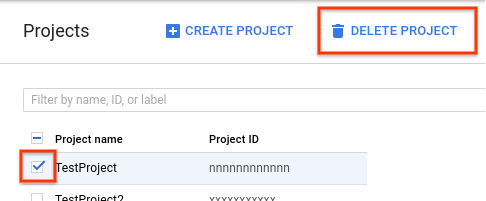
- In the dialog, type the project ID, and then click Shut down to delete the project.
What's next
- If you have questions about the pipeline or run into issues, email support@sentieon.com.