Ce tutoriel vous montre comment prétraiter, entraîner et effectuer des prédictions avec un modèle de machine learning à l'aide d'Apache Beam, de Google Dataflow et de TensorFlow.
Pour illustrer ces concepts, cette procédure utilise l'exemple de code Molecules. À partir de données moléculaires en entrée, le code Molecules crée et entraîne un modèle d'apprentissage automatique permettant de prédire l'énergie moléculaire.
Coûts
Ce tutoriel peut utiliser des composants facturables de Google Cloud, dont un ou plusieurs des éléments suivants :
- Dataflow
- Cloud Storage
- AI Platform
Obtenez une estimation des coûts en fonction de votre utilisation prévue à l'aide du simulateur de coût.
Présentation
Le code Molecules extrait des fichiers contenant des données moléculaires, et compte le nombre d'atomes de carbone, d'hydrogène, d'oxygène et d'azote dans chaque molécule. Ensuite, le code normalise les décomptes vers des valeurs comprises entre 0 et 1, et transmet ces valeurs à un Estimator de réseau de neurones profond TensorFlow. L'Estimator de réseau de neurones entraîne un modèle de machine learning pour prédire l'énergie moléculaire.
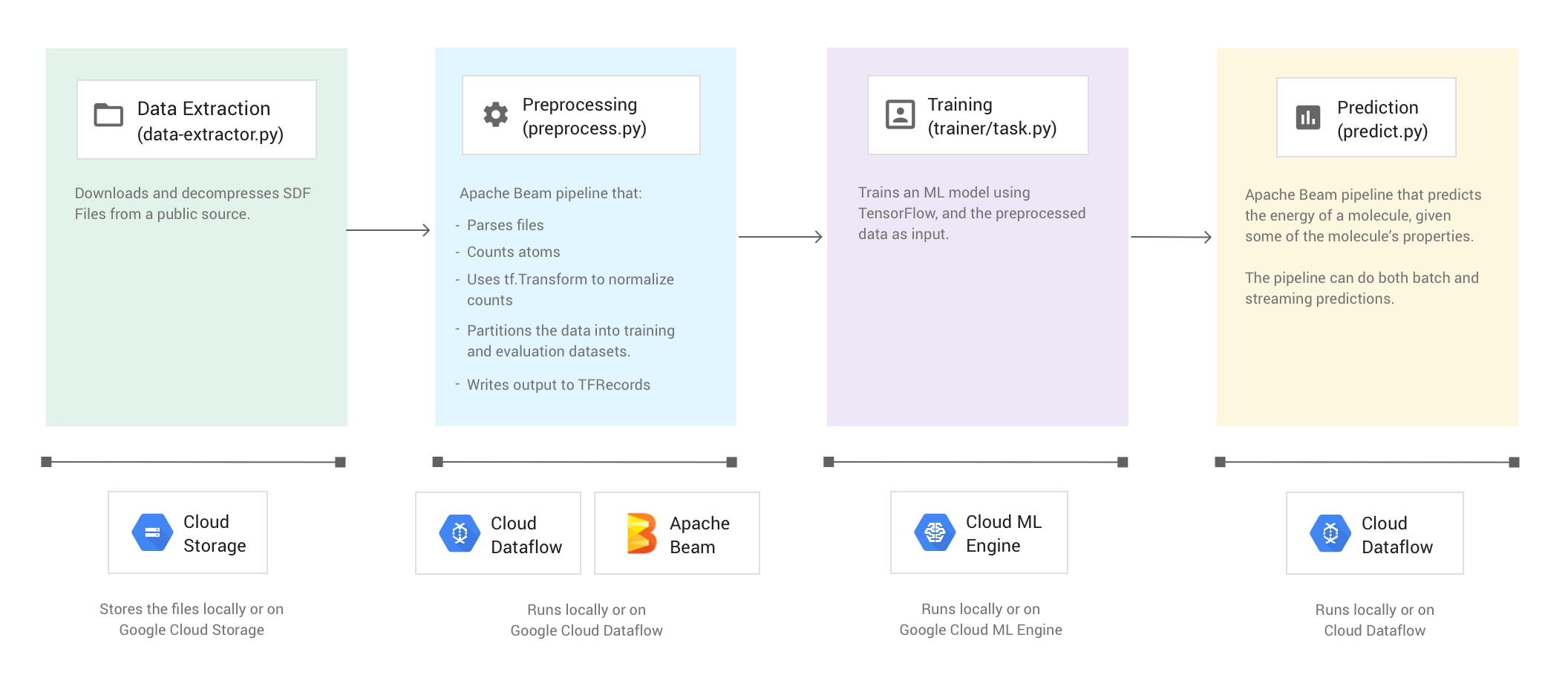
L'exemple de code est organisé en quatre phases :
- Extraction de données (
data-extractor.py) - Prétraitement (
preprocess.py) - Entraînement (
trainer/task.py) - Prédiction (
predict.py)
Les sections ci-dessous décrivent les quatre phases, mais ce tutoriel porte principalement sur les phases qui utilisent Apache Beam et Dataflow : la phase de prétraitement et la phase de prédiction. La phase de prétraitement utilise également la bibliothèque TensorFlow Transform (communément appelée tf.Transform).
L'illustration suivante montre le workflow de l'exemple de code Molecules.
Exécuter l'exemple de code
Pour configurer votre environnement, suivez les instructions figurant dans le fichier README du dépôt GitHub de Molecules. Exécutez ensuite l'exemple de code Molecules à l'aide de l'un des scripts wrapper fournis, run-local ou run-cloud. Ces scripts exécutent automatiquement les quatre phases de l'exemple de code (extraction, prétraitement, entraînement et prédiction).
Vous pouvez également exécuter manuellement chacune des phases à l'aide des commandes fournies dans les sections de ce document.
Exécuter localement
Pour exécuter l'exemple de code Molecules en local, exécutez le script wrapper run-local :
./run-local
Les journaux de sortie vous indiquent laquelle des quatre phases le script est en train d'exécuter (extraction de données, prétraitement, entraînement et prédiction).
Le script data-extractor.py requiert un argument correspondant au nombre de fichiers.
Pour faciliter leur utilisation, les scripts run-local et run-cloud possèdent une valeur par défaut de cinq fichiers pour cet argument. Chaque fichier contient 25 000 molécules. Exécuter cet exemple de code doit prendre approximativement de trois à sept minutes du début à la fin. La durée d'exécution dépend du processeur de votre ordinateur.
Exécuter sur Google Cloud
Pour exécuter l'exemple de code Molecules sur Google Cloud, exécutez le script wrapper run-cloud. Tous les fichiers d'entrée doivent être hébergés dans Cloud Storage.
Définissez le paramètre --work-dir pour qu'il pointe vers le bucket Cloud Storage :
./run-cloud --work-dir gs://<your-bucket-name>/cloudml-samples/molecules
Phase 1 : Extraction des données
Code source : data-extractor.py
La première étape consiste à extraire les données d'entrée. Le fichier data-extractor.py extrait et décompresse les fichiers SDF spécifiés. Au cours d'étapes suivantes, l'exemple prétraite ces fichiers, et utilise les données pour entraîner et évaluer le modèle de machine learning. Le script extrait les fichiers SDF de leur source publique et les enregistre dans un sous-répertoire du répertoire de travail spécifié. Le répertoire de travail par défaut (--work-dir) est /tmp/cloudml-samples/molecules.
Enregistrer les fichiers extraits
Enregistrez localement les fichiers de données que vous venez d'extraire :
python data-extractor.py --max-data-files 5
Ou enregistrez les fichiers extraits dans un emplacement Cloud Storage :
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python data-extractor.py --work-dir $WORK_DIR --max-data-files 5
Phase 2 : Prétraitement
Code source : preprocess.py
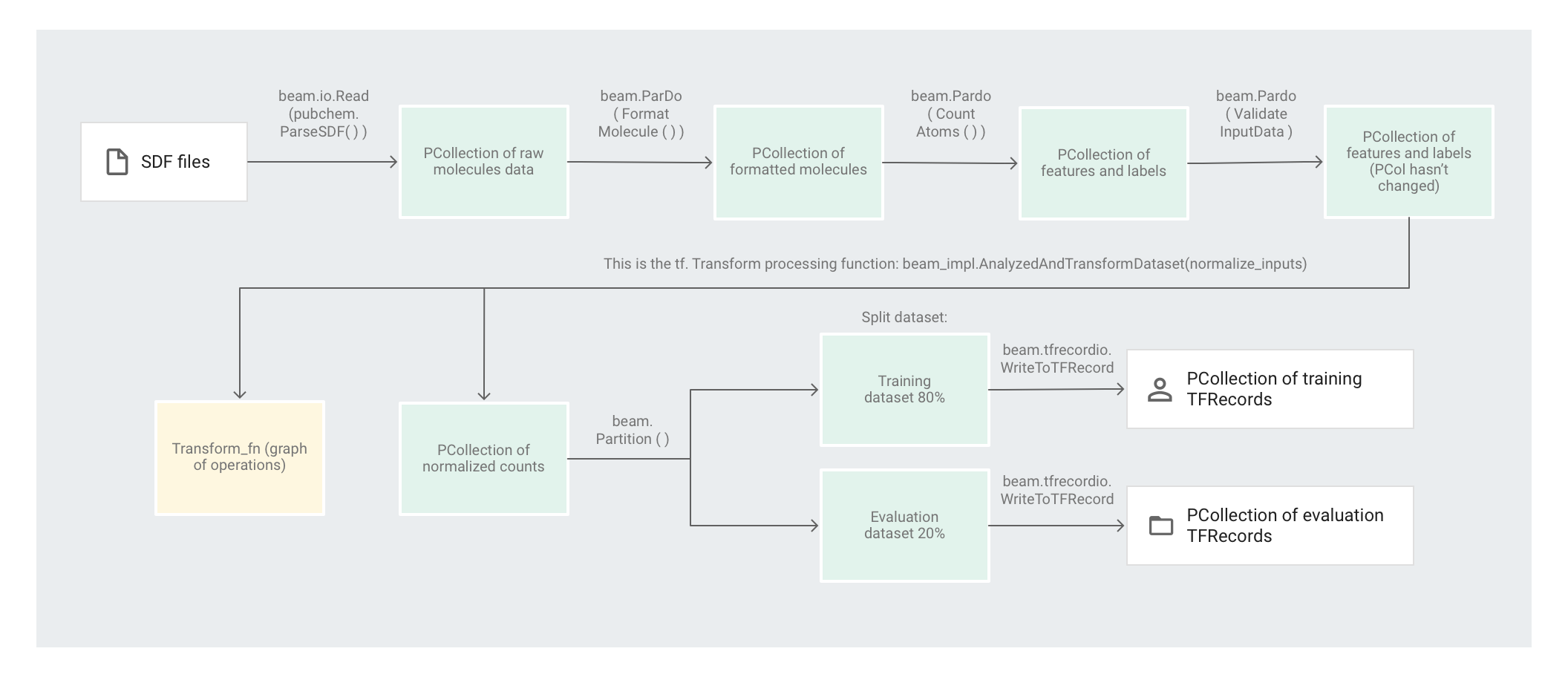
L'exemple de code Molecules utilise un pipeline Apache Beam pour prétraiter les données. Le pipeline effectue les opérations de prétraitement suivantes :
- Lire et analyser les fichiers SDF extraits.
- Compter le nombre d'atomes différents dans chacune des molécules des fichiers.
- Normaliser les décomptes à l'aide de
tf.Transformvers des valeurs comprises entre 0 et 1. - Partitionner l'ensemble de données en un ensemble de données d'apprentissage et un ensemble de données d'évaluation.
- Écrire les deux ensembles de données en tant qu'objets
TFRecord.
Les transformations Apache Beam peuvent manipuler efficacement des éléments individuels, mais les transformations impliquant de parcourir l'intégralité de l'ensemble de données sont difficiles à réaliser avec Apache Beam uniquement. Elles sont plus efficaces avec tf.Transform. De ce fait, le code utilise des transformations Apache Beam pour lire et mettre en forme les molécules, et pour compter les atomes dans chaque molécule. Le code utilise ensuite tf.Transform pour identifier les nombres minimum et maximum globaux afin de normaliser les données.
L'illustration suivante montre les étapes du pipeline.
Appliquer des transformations basées sur les éléments
Le code preprocess.py crée un pipeline Apache Beam.
Ensuite, le code applique une transformation feature_extraction au pipeline.
Le pipeline utilise SimpleFeatureExtraction en tant que transformation feature_extraction.
La transformation SimpleFeatureExtraction, définie dans pubchem/pipeline.py, contient une série de transformations qui manipulent tous les éléments indépendamment.
Pour commencer, le code analyse les molécules à partir du fichier source, puis il met en forme les molécules dans un dictionnaire des propriétés moléculaires et enfin, il compte les atomes présents dans la molécule. Ces nombres sont les caractéristiques (entrées) du modèle de machine learning.
La transformation de lecture beam.io.Read(pubchem.ParseSDF(data_files_pattern)) lit les fichiers SDF depuis une source personnalisée.
La source personnalisée, appelée ParseSDF, est définie dans pubchem/pipeline.py.
ParseSDF étend FileBasedSource et implémente la fonction read_records qui ouvre les fichiers SDF extraits.
Lorsque vous exécutez l'exemple de code Molecules sur Google Cloud, plusieurs nœuds de calcul (VM) peuvent lire simultanément les fichiers. Pour éviter que deux nœuds de calcul ne lisent le même contenu dans les fichiers, chaque fichier utilise une valeur range_tracker.
Le pipeline regroupe les données brutes en sections d'informations pertinentes nécessaires aux étapes suivantes. Chaque section du fichier SDF analysé est stockée dans un dictionnaire (voir pipeline/sdf.py), dans lequel les clés sont les noms des sections et les valeurs correspondent au contenu brut des lignes de la section correspondante.
Le code applique beam.ParDo(FormatMolecule()) au pipeline. La fonction ParDo applique le DoFn nommé FormatMolecule à chaque molécule. FormatMolecule génère un dictionnaire de molécules mises en forme. Le fragment de code suivant est un exemple d'élément figurant dans la PCollection de sortie :
{
'atoms': [
{
'atom_atom_mapping_number': 0,
'atom_stereo_parity': 0,
'atom_symbol': u'O',
'charge': 0,
'exact_change_flag': 0,
'h0_designator': 0,
'hydrogen_count': 0,
'inversion_retention': 0,
'mass_difference': 0,
'stereo_care_box': 0,
'valence': 0,
'x': -0.0782,
'y': -1.5651,
'z': 1.3894,
},
...
],
'bonds': [
{
'bond_stereo': 0,
'bond_topology': 0,
'bond_type': 1,
'first_atom_number': 1,
'reacting_center_status': 0,
'second_atom_number': 5,
},
...
],
'<PUBCHEM_COMPOUND_CID>': ['3\n'],
...
'<PUBCHEM_MMFF94_ENERGY>': ['19.4085\n'],
...
}
Ensuite, le code applique beam.ParDo(CountAtoms()) au pipeline. Le DoFnCountAtoms comptabilise le nombre d'atomes de carbone, d'hydrogène, d'azote et d'oxygène contenus dans chaque molécule. CountAtoms renvoie une PCollection de caractéristiques et de libellés.
Voici un exemple d'élément figurant dans la PCollection de sortie :
{
'ID': 3,
'TotalC': 7,
'TotalH': 8,
'TotalO': 4,
'TotalN': 0,
'Energy': 19.4085,
}
Le pipeline valide ensuite les entrées. Le ValidateInputDataDoFn valide le fait que chaque élément correspond aux métadonnées renseignées dans le input_schema. Cette validation garantit le bon format des données lors de leur transfert à TensorFlow.
Appliquer des transformations effectuant un passage complet
L'exemple de code Molecules utilise un régresseur de réseau de neurones profonds pour effectuer des prédictions. Il est généralement recommandé de normaliser les entrées avant de les introduire dans le modèle de ML. Le pipeline utilise tf.Transform pour normaliser les décomptes de chaque atome vers des valeurs comprises entre 0 et 1. Pour en savoir plus sur la normalisation des entrées, consultez l'article Feature scaling (Scaling des caractéristiques).
La normalisation des valeurs implique un passage complet sur l'ensemble de données pour enregistrer les valeurs minimale et maximale. Le code utilise tf.Transform pour parcourir l'intégralité de l'ensemble de données et appliquer des transformations en passage complet.
Pour utiliser tf.Transform, le code doit fournir une fonction contenant la logique de la transformation à effectuer sur l'ensemble de données. Dans preprocess.py, le code utilise la transformation AnalyzeAndTransformDataset fournie par tf.Transform. En savoir plus sur l'utilisation de tf.Transform.
Dans preprocess.py, la fonction de feature_scaling utilisée est normalize_inputs. Elle est définie dans pubchem/pipeline.py. La fonction utilise la fonction tf.Transform scale_to_0_1 pour normaliser les décomptes vers des valeurs comprises entre 0 et 1.
Il est possible de normaliser manuellement les données, mais si l'ensemble de données est volumineux, il est plus rapide d'utiliser Dataflow. L'utilisation de Dataflow permet au pipeline de s'exécuter sur plusieurs nœuds de calcul (VM) si nécessaire.
Partitionner l'ensemble de données
Ensuite, le pipeline preprocess.py partitionne l'ensemble de données unique en deux ensembles de données distincts. Il alloue environ 80 % des données à l'entraînement et environ 20 % à l'évaluation.
Écrire la sortie
Enfin, le pipeline preprocess.py écrit les deux ensembles de données (entraînement et évaluation) à l'aide de la transformation WriteToTFRecord.
Exécuter le pipeline de prétraitement
Exécutez le pipeline de prétraitement localement :
python preprocess.py
Vous pouvez également exécuter le pipeline de prétraitement sur Dataflow :
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python preprocess.py \
--project $PROJECT \
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--work-dir $WORK_DIR
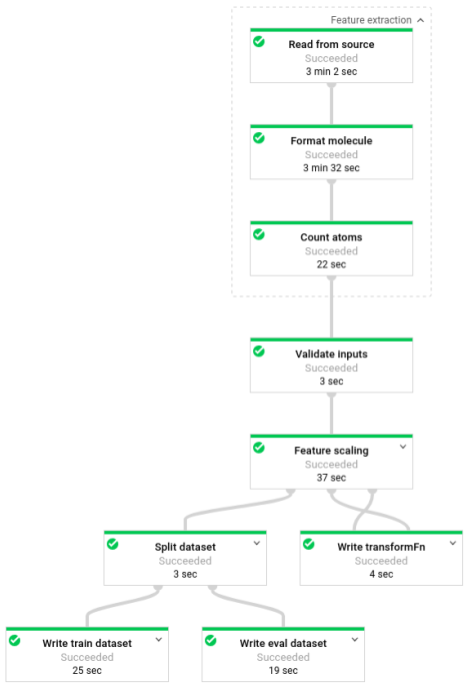
Une fois le pipeline en cours d'exécution, vous pouvez afficher sa progression dans l'interface de surveillance de Dataflow :
Phase 3 : Entraînement
Code source : trainer/task.py
Rappelons qu'à la fin de la phase de prétraitement, le code partitionne les données en deux ensembles de données (entraînement et évaluation).
L'exemple utilise TensorFlow pour entraîner le modèle de machine learning. Le fichier trainer/task.py de l'exemple Molecules contient le code d'entraînement du modèle. La fonction principale de trainer/task.py charge les données traitées lors de la phase de prétraitement.
L'Estimator utilise l'ensemble de données d'apprentissage pour entraîner le modèle, puis l'ensemble de données d'évaluation pour vérifier que le modèle prédit avec justesse l'énergie moléculaire en fonction de certaines propriétés de la molécule.
En savoir plus sur l'entraînement d'un modèle de ML.
Entraîner le modèle
Entraînez le modèle localement :
python trainer/task.py
# To get the path of the trained model
EXPORT_DIR=/tmp/cloudml-samples/molecules/model/export/final
MODEL_DIR=$(ls -d -1 $EXPORT_DIR/* | sort -r | head -n 1)
Sinon, entraînez le modèle sur AI Platform :
- Sélectionnez d'abord une région compatible pour exécuter la tâche d'entraînement
gcloud config set compute/region $REGION
- Lancez la tâche d'entraînement
JOB="cloudml_samples_molecules_$(date +%Y%m%d_%H%M%S)"
BUCKET=gs://<your bucket name>
WORK_DIR=$BUCKET/cloudml-samples/molecules
gcloud ai-platform jobs submit training $JOB \
--module-name trainer.task \
--package-path trainer \
--staging-bucket $BUCKET \
--runtime-version 1.13 \
--stream-logs \
-- \
--work-dir $WORK_DIR
# To get the path of the trained model:
EXPORT_DIR=$WORK_DIR/model/export
MODEL_DIR=$(gsutil ls -d $EXPORT_DIR/* | sort -r | head -n 1)
Phase 4 : Prédiction
Code source : predict.py
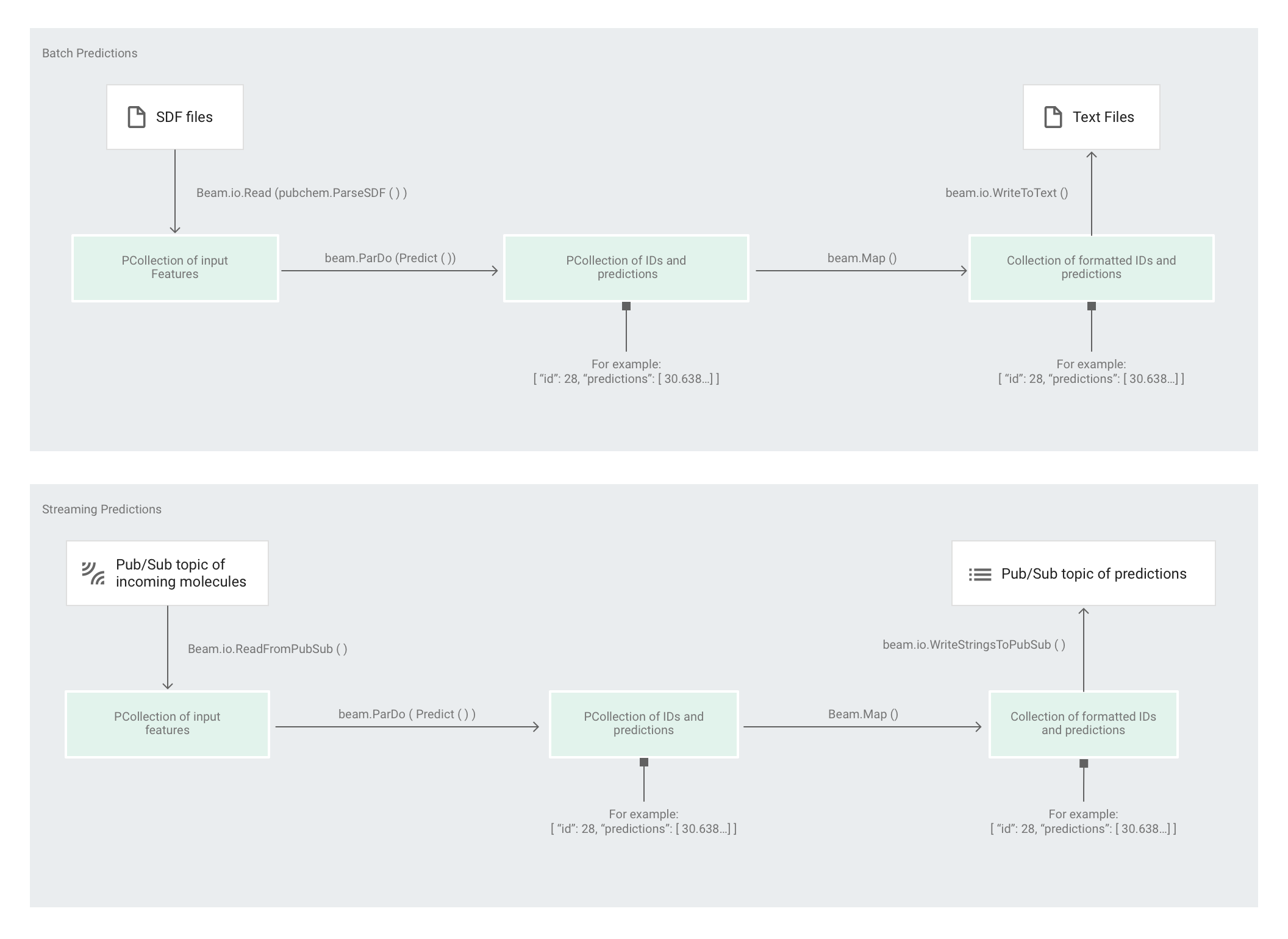
Une fois que l'Estimator a entraîné le modèle, vous pouvez lui fournir des données d'entrée qui lui permettront de faire des prédictions. Dans l'exemple de code Molecules, c'est le pipeline de predict.py qui est chargé d'effectuer les prédictions. Le pipeline peut fonctionner par lots ou par flux.
Le code du pipeline est le même pour le traitement par lots et par flux, à l'exception des interactions source et récepteur. Si le pipeline s'exécute en mode de traitement par lots, il lit les fichiers d'entrée à partir de la source personnalisée et écrit les prédictions de sortie sous forme de fichiers texte dans le répertoire de travail spécifié. Si le pipeline s'exécute en mode de traitement par flux, il lit les molécules d'entrée, à mesure qu'elles arrivent, à partir d'un sujet Pub/Sub. Lorsque les prédictions de sortie sont prêtes, le pipeline les écrit dans un autre sujet Pub/Sub.
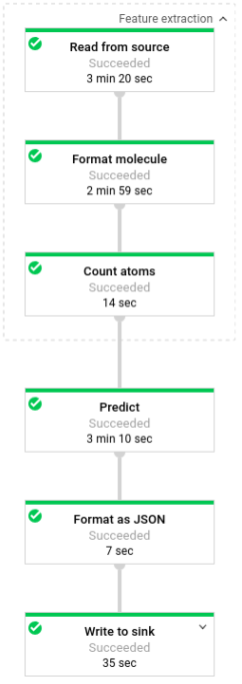
L'illustration suivante montre les étapes des pipelines de prédiction (par lots et par flux).
Dans predict.py, le code définit le pipeline dans la fonction run :
Le code appelle la fonction run avec les paramètres suivants :
Tout d'abord, le code transmet la transformation pubchem.SimpleFeatureExtraction(source) en tant que transformation feature_extraction. Cette transformation, également utilisée dans la phase de prétraitement, est appliquée au pipeline :
La transformation lit les données dans la source adéquate suivant le mode d'exécution du pipeline (par lot ou par flux), met en forme les molécules et compte les différents atomes dans chaque molécule.
Ensuite, la fonction beam.ParDo(Predict(…)) est appliquée au pipeline chargé de prédire l'énergie moléculaire.
Predict, la fonction DoFn qui est transmise, utilise le dictionnaire des caractéristiques d'entrée (décompte des atomes) pour prédire l'énergie moléculaire.
La transformation suivante appliquée au pipeline est beam.Map(lambda result: json.dumps(result)), qui prend le dictionnaire de résultats de prédiction et le sérialise en une chaîne JSON.
Enfin, la sortie est écrite dans le récepteur (sous forme de fichiers texte dans le répertoire de travail pour le traitement par lot ou sous forme de messages publiés dans un sujet Pub/Sub pour le traitement par flux).
Prédictions par lots
Les prédictions par lots sont optimisées pour le débit plutôt que pour la latence. Elles fonctionnent mieux si vous devez effectuer un certain nombre de prédictions, mais que vous pouvez attendre qu'elles soient toutes disponibles avant d'obtenir les résultats.
Exécuter le pipeline de prédiction en mode de traitement par lots
Exécutez le pipeline de prédiction par lots localement :
# For simplicity, we'll use the same files we used for training
python predict.py \
--model-dir $MODEL_DIR \
batch \
--inputs-dir /tmp/cloudml-samples/molecules/data \
--outputs-dir /tmp/cloudml-samples/molecules/predictions
Vous pouvez également exécuter le pipeline de prédiction par lots sur Dataflow :
# For simplicity, we'll use the same files we used for training
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python predict.py \
--work-dir $WORK_DIR \
--model-dir $MODEL_DIR \
batch \
--project $PROJECT \
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--inputs-dir $WORK_DIR/data \
--outputs-dir $WORK_DIR/predictions
Une fois le pipeline en cours d'exécution, vous pouvez afficher sa progression dans l'interface de surveillance de Dataflow :
Prédictions par flux
Les prédictions par flux sont optimisées pour la latence plutôt que pour le débit. Elles fonctionnent mieux si vous effectuez des prédictions de façon sporadique, mais que vous souhaitez obtenir les résultats le plus rapidement possible.
Le service de prédiction (pipeline de prédiction par flux) reçoit les molécules d'entrée depuis un sujet Pub/Sub et publie la sortie (les prédictions) dans un autre sujet Pub/Sub.
Créez le sujet Pub/Sub pour les entrées :
gcloud pubsub topics create molecules-inputs
Créez le sujet Pub/Sub pour les sorties :
gcloud pubsub topics create molecules-predictions
Exécutez le pipeline de prédiction par flux localement :
# Run on terminal 1
PROJECT=$(gcloud config get-value project)
python predict.py \
--model-dir $MODEL_DIR \
stream \
--project $PROJECT
--inputs-topic molecules-inputs \
--outputs-topic molecules-predictions
Vous pouvez également exécuter le pipeline de prédiction par flux sur Dataflow :
# Run on terminal 1
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python predict.py \
--work-dir $WORK_DIR \
--model-dir $MODEL_DIR \
stream \
--project $PROJECT
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--inputs-topic molecules-inputs \
--outputs-topic molecules-predictions
Une fois que le service de prédiction (le pipeline de prédiction par flux) est fonctionnel, vous devez exécuter un éditeur pour envoyer les molécules au service de prédiction et un abonné pour écouter les résultats de la prédiction. L'exemple de code Molecules fournit le code pour les services d'éditeur (publisher.py) et d'abonné (subscriber.py).
L'éditeur analyse les fichiers SDF depuis un répertoire et les publie dans le sujet des entrées. L'abonné écoute les résultats de la prédiction et les enregistre. Pour des raisons de simplicité, cet exemple utilise les mêmes fichiers SDF que ceux utilisés pour la phase d'entraînement.
Exécutez l'abonné :
# Run on terminal 2
python subscriber.py \
--project $PROJECT \
--topic molecules-predictions
Exécutez l'éditeur :
# Run on terminal 3
python publisher.py \
--project $PROJECT \
--topic molecules-inputs \
--inputs-dir $WORK_DIR/data
Une fois que l'éditeur commence à analyser et à publier des molécules, vous commencez à voir apparaître les prédictions au niveau de l'abonné.
Effectuer un nettoyage
Dès que vous avez terminé d'exécuter le pipeline de prédictions par flux, arrêtez votre pipeline pour éviter les frais.
Étapes suivantes
- Consultez la documentation Apache Beam.
- Consultez la documentation TensorFlow :
- Essayez un autre exemple de code utilisant Apache Beam et
tf.Transform. - Entraînez un modèle de ML pour la classification de séries temporelles à l'aide de l'exemple Global Fishing Watch.