BigQuery と Vertex AI を使用した RNA シーケンスとタンパク質構造予測

Google Cloud Japan Team
※この投稿は米国時間 2023 年 6 月 22 日に、Google Cloud blog に投稿されたものの抄訳です。
RNA シーケンスとタンパク質構造予測は現代の生物学的研究に不可欠なツールであり、疾患の分子機序を明らかにして有望な治療法の開発につなげる知見の発見を促進します。RNA シーケンスは遺伝子発現をプロファイリングするための手法で、遺伝子調節や遺伝子間の複雑な相互作用の理解を深めるのに役立ちます。それに対してタンパク質構造予測は、タンパク質の機能や他の分子との相互作用に関する情報を提供します。これは医薬品開発に有効であり、標的結合部位の特定や新薬候補の最適化を可能にします。
課題
これらの手法によって生成される膨大な量のデータを扱うのは複雑な作業で、この複雑さが研究者にとって大きな課題となっています。RNA シーケンスとタンパク質構造予測において正確な解析を行うには、かなりの計算オーバーヘッドとストレージ オーバーヘッドが必要になります。タンパク質構造予測は、タンパク質の大きさや多数のタンパク質の複雑な結合ダイナミクスによってさらに複雑化され、それが正確な予測を困難にしています。また、多くのタンパク質には正解とされる構造データが欠如しているため、計算による予測の妥当性検証も骨が折れます。その結果、タスクの複雑さがさらに増しています。こうした課題を解決するには、正確で信頼性の高い結果を達成するスケーラブルで再現可能な解析アプローチが必要です。
ソリューション
近年の ML とクラウド コンピューティングの進歩は、このような課題に取り組む新たな道を切り開きました。AlphaFold(DeepMind が開発したディープ ラーニング プログラム)は、入力したアミノ酸配列からタンパク質の 3D 構造や他の分子との結合ダイナミクスを正確に予測できます。Nextflow(オープンソースのワークフロー管理システム)と Google Batch(クラウド コンピューティング リソース上でのバッチジョブのスケジューリング、キューイング、実行を担うフルマネージド サービス)を使用すると、Docker コンテナを利用してスケーラブルかつ再現可能な形でゲノムデータを解析できます。私たちは RNA シーケンスとタンパク質構造予測に Google Cloud の力を活用できないかと考え、BigQuery と Vertex AI を利用してテラバイト規模のデータを効率的に処理するエンドツーエンドのパイプラインを開発しました。ここでは、このパイプラインの開発について詳しく説明します。この記事が、Google Cloud を使用して現代の生物学や医学が抱えるコンピューティングの課題に立ち向かうための有益なヒントを与え、最終的に新たな発見やイノベーションへの道筋をつける助けになれば幸いです。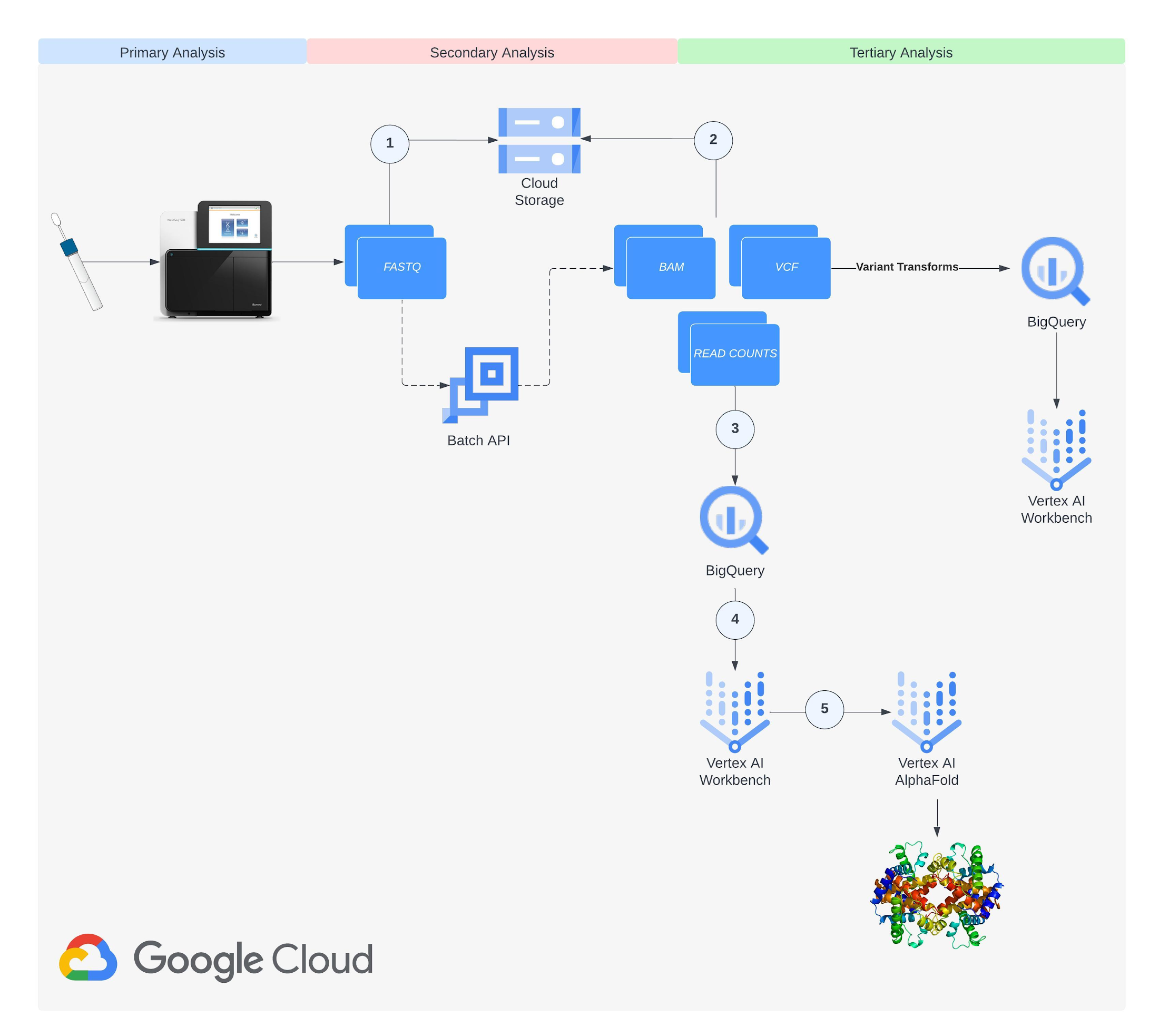
方法
ステップ 1: 準備
Cloud Storage バケットを作成します。バケットの作成方法については、こちらのドキュメントをご覧ください。プロジェクトで Artifact Registry、Batch、Dataflow、Cloud Life Sciences、Compute Engine、Notebooks、Cloud Storage、Vertex AI の API を有効にします。サービス アカウントを作成し、Cloud Life Sciences ワークフロー実行者、サービス アカウント ユーザー、Service Usage ユーザー、ストレージ オブジェクト管理者の各ロールをプロジェクト レベルで追加します。サービス アカウントの作成方法については、こちらのドキュメントをご覧ください。
ステップ 2: データ収集
次のステップでは、「Single-cell profiling of human bone marrow progenitors reveals mechanisms of failing erythropoiesis in Diamond-Blackfan anemia」という論文を原典とする NCBI の一般公開データセット(GSE181830)からデータを収集します。この研究で使用された患者メタデータと FASTQ ファイル(*.fq.gz)は NCBI の SRA Run Selector サービスからダウンロード可能で、指定のバケットに設定した NCBI のサービス アカウントにストレージ オブジェクト管理者のアクセス権を一時的に付与することで Cloud Storage に直接取り込むことができます。データの取り込みが完了したら、確認メールが届きます。これで、選択したファイルが指定のバケットに格納されます。別の方法として、European Nucleotide Archive(ENA)ウェブサイトから FASTQ ファイルをダウンロードすることもできます。この場合は、各ランの SRA ID を入力し、wget または curl を使用してファイルをローカル ディレクトリにダウンロードしてから、gsutil を使用してそれらのファイルを Cloud Storage バケットにコピーします。
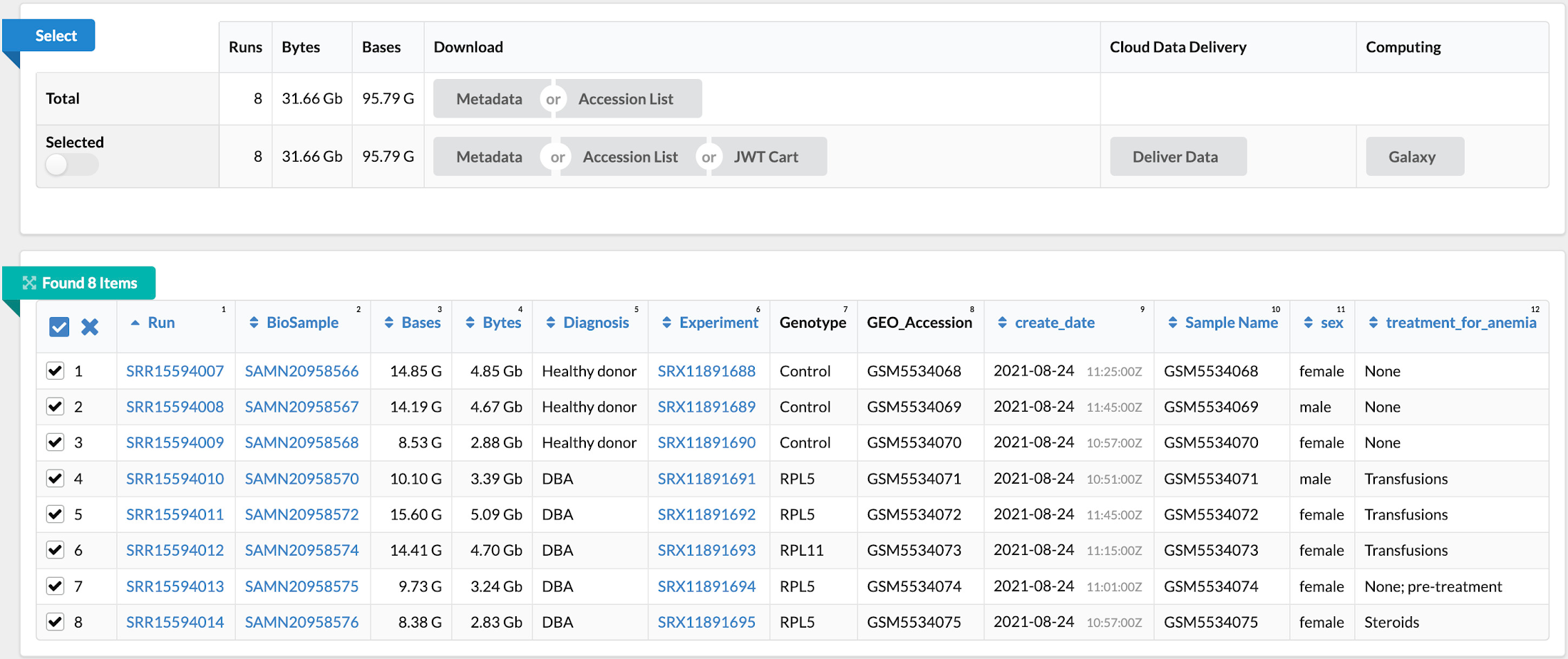
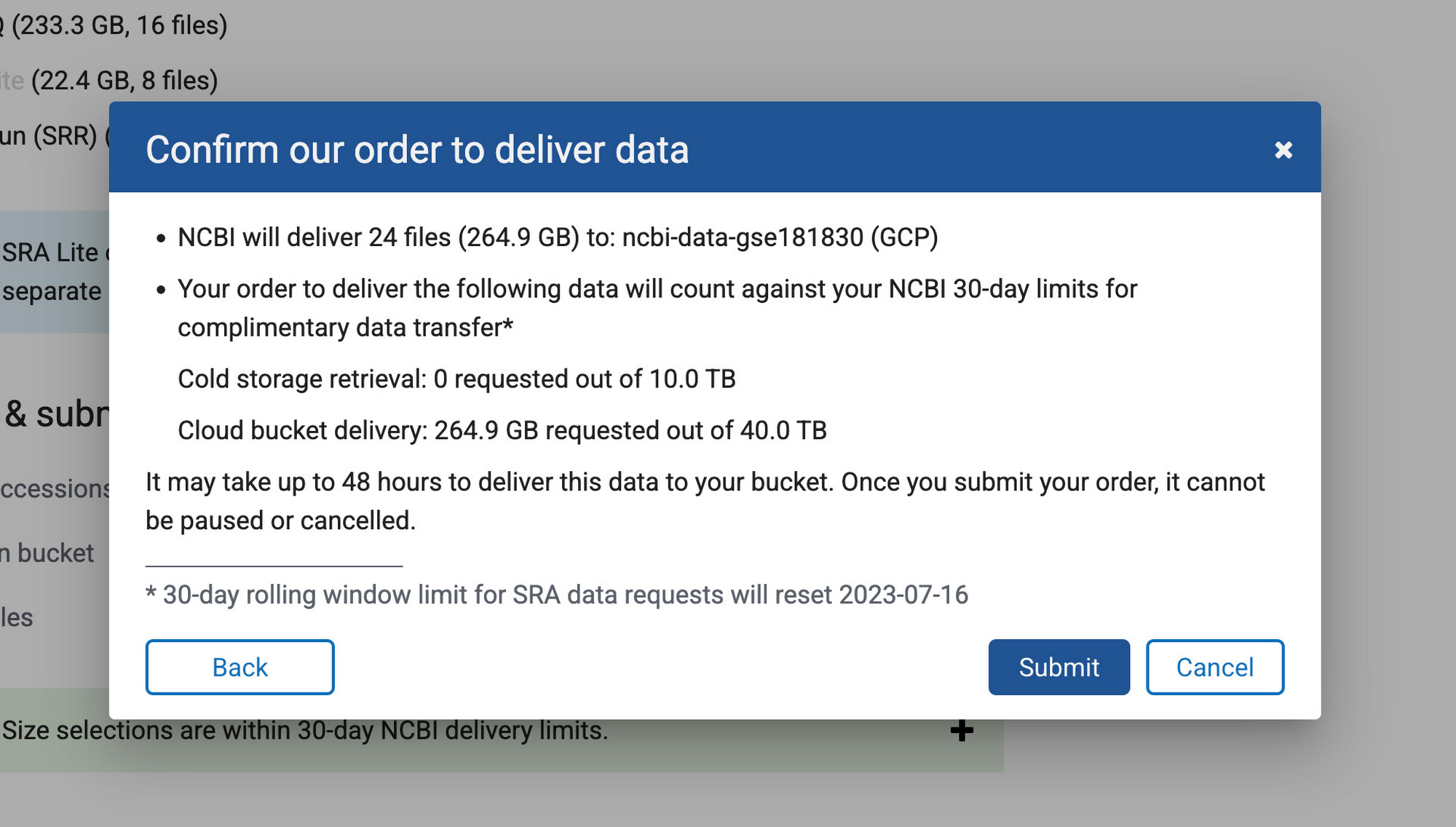
ステップ 3: ゲノム インデックスを構築する
次のステップでは、RNA シーケンス解析のアライメント ステップでリードのマッピングに使用されるゲノム インデックス(GRCh38.v42)を作成します。Vertex AI Workbench のユーザー管理ノートブックで、Gencode からダウンロードして Cloud Storage にアップロードした転写物およびアノテーション ファイルと、GitHub からダウンロード可能なオープンソースの STAR RNA アライナー ライブラリ(2.7.10c)を使用して、ゲノム インデックスを構築します。ユーザー管理ノートブック インスタンスの作成方法については、こちらのドキュメントをご覧ください。指定の出力ディレクトリにゲノム インデックスを作成する処理には約 1 時間かかります。完了したら、ステップ 1 で作成した Cloud Storage バケットに出力ディレクトリをアップロードします。
ステップ 4: コンテナ イメージをビルドして Artifact Registry にアップロードする
次のステップでは、Docker コンテナ イメージをビルドし、Google Cloud のアーティファクト リポジトリである Artifact Registry にアップロードします。Nextflow は、ユーザーが指定したコンテナでパイプラインの各ステップを実行します。パイプラインで使用するイメージをビルドし、fastqc、multiqc、rna-star、rsem やパイプラインで参照する BigQuery Python クライアント ライブラリなどの依存関係やライブラリとともにイメージを読み込む必要があります。リポジトリを作成する方法についてはこちらを、Artifact Registry にイメージを push および pull する方法についてはこちらを、それぞれご覧ください。イメージを作成してリポジトリに push したら、ステップ 1 で作成したサービス アカウントに Artifact Registry 読み取りのロールを付与します。今回のワークフローで使用した Dockerfile は GitHub で公開されています。
ステップ 5: 遺伝子およびアイソフォーム発現データ用の BigQuery データセットを作成する
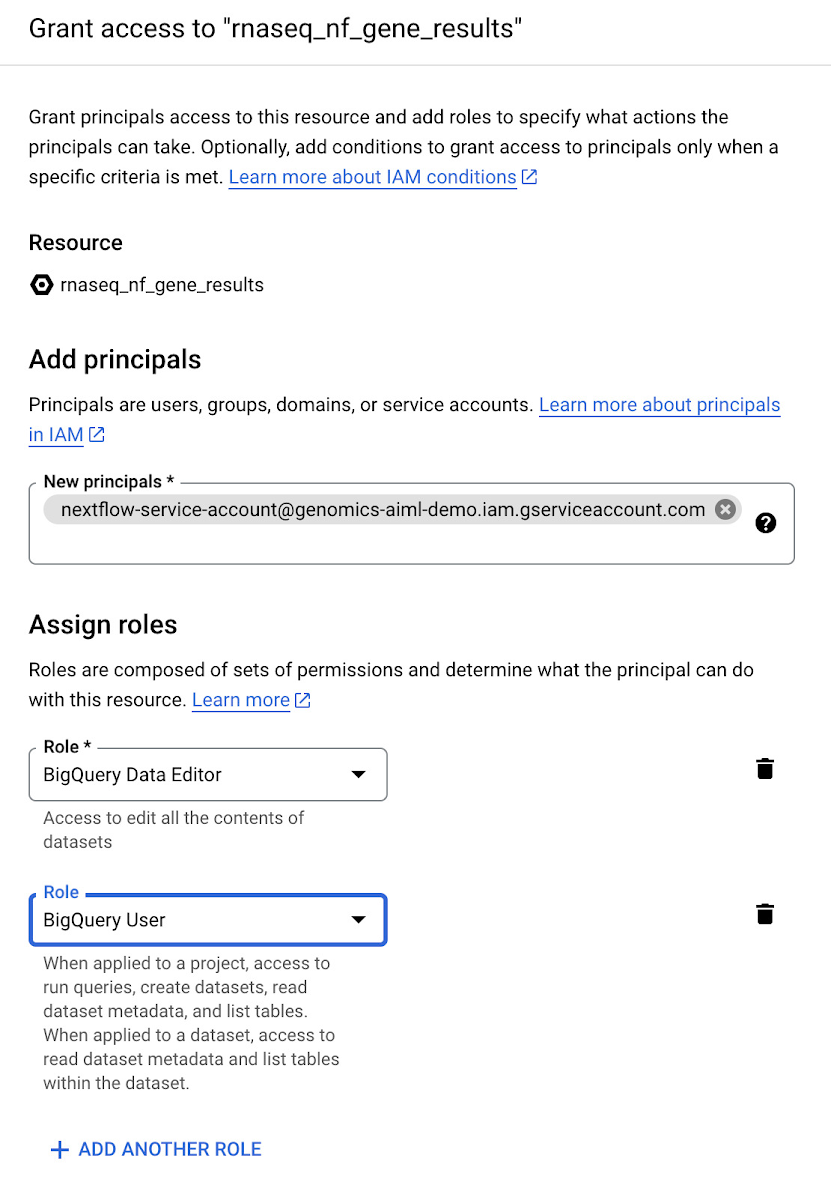
次のステップでは、遺伝子とアイソフォームのリード数データを格納する BigQuery データセットを作成します。RNA シーケンス パイプラインの最後のステップでリード数データを BigQuery テーブルに書き込むため、これらのデータセットを事前に作成しておく必要があります。BigQuery でデータセットを作成する方法については、こちらのドキュメントをご覧ください。最初に作成するデータセットの名前は rnaseq_nf_gene_results とし、2 つ目のデータセットの名前は rnaseq_nf_isoform_results とします。両方のデータセットで、ステップ 1 で作成したサービス アカウントに BigQuery エディタと BigQuery ユーザーのロールを付与します。
ステップ 6: Nextflow のパイプラインを構成する
次のステップでは、ペアエンド RNA シーケンス前処理ワークフローを実行する Nextflow のパイプラインを構成します。そのためには、nextflow.config ファイルと main.nf ファイルを GitHub リポジトリに作成する必要があります。今回のワークフローで使用した nextflow.config、main.nf、load_rsem_results_into_bq.py(main.nf 内で参照されている)の各ファイルは GitHub で公開されています。Nextflow スクリプトの作成については、こちらのドキュメントをご覧ください。nextflow.config スクリプトに含まれるプロジェクトやバケットの設定を、実際に使用する設定に置き換えます。main.nf スクリプトに記述されているパイプラインのステップは次のとおりです。(1)Trim Galore によるアダプタとクオリティのトリミング、(2)FastQC による読み取り値の品質管理、(3)RSEM による遺伝子およびアイソフォーム発現のアライメントと推定、(4)発現データの BigQuery への書き込み。
ステップ 7: パイプラインを Nextflow Tower にデプロイする
次のステップでは、パイプラインを Nextflow Tower にデプロイします。Tower は、サービス アカウント キーを介してパイプラインを実行する Google プロジェクトへの認証をサポートしています。サービス アカウント キーを接続する方法については、こちらのドキュメントをご覧ください。Tower は、アクセス トークンを使用して Nextflow 構成をホストする Git リポジトリへの接続もサポートしています。Tower でリポジトリを接続する方法については、こちらのドキュメントをご覧ください。サービス アカウントとリポジトリの認証情報を設定したら、次にコンピューティング環境を作成します。[Add Compute Environment] ビューで、コンピューティング環境につける任意の名前(例: RNASeq-NF-GCP)を入力し、[Platform] プルダウンで Google Batch を選択します。次に、ワークロードを作成するリージョンを入力し(ステップ 6 で nextflow.config に指定したのと同じリージョンを使用できます)、パイプラインの作業ディレクトリ(GCS のパス)を入力します。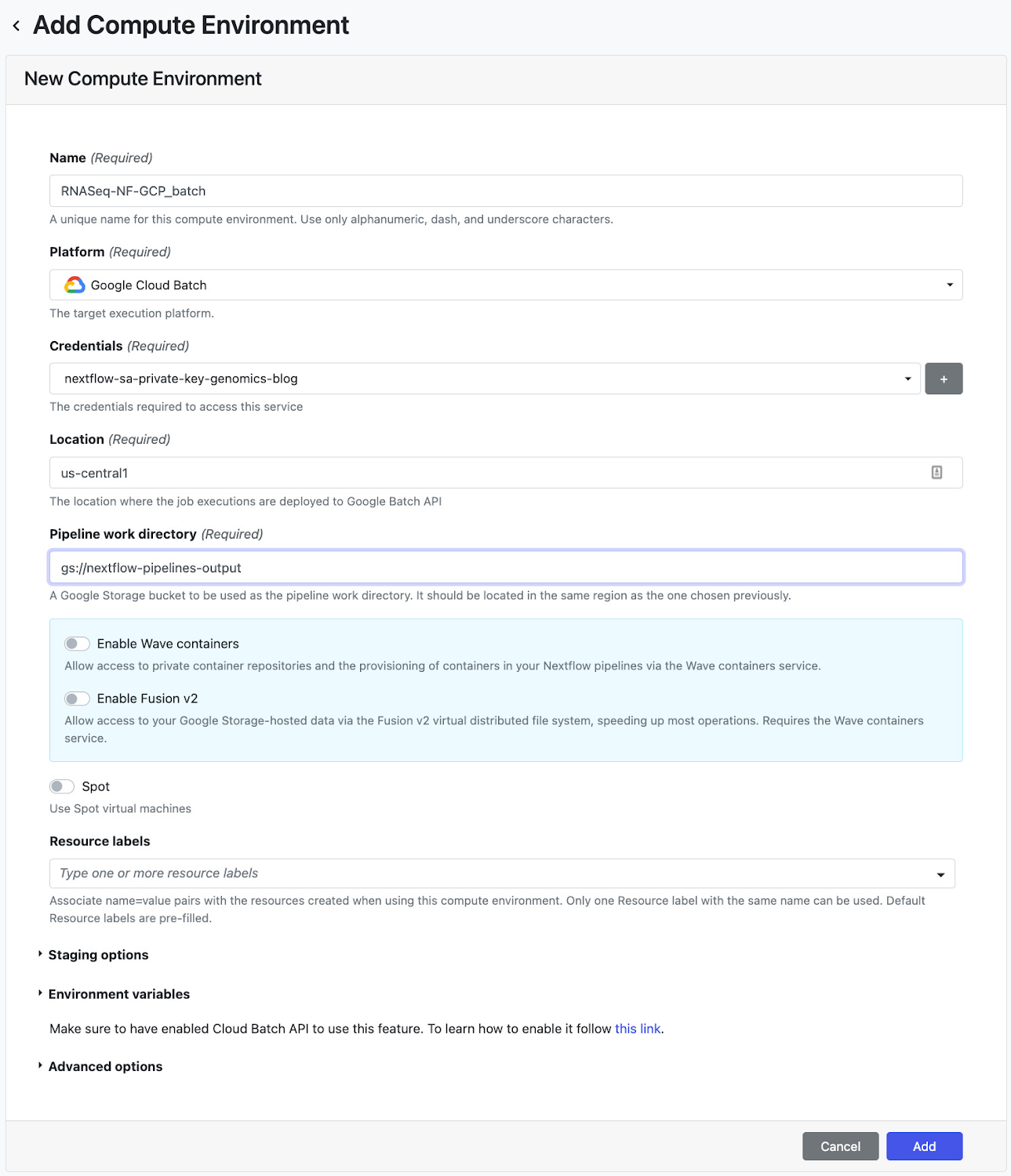
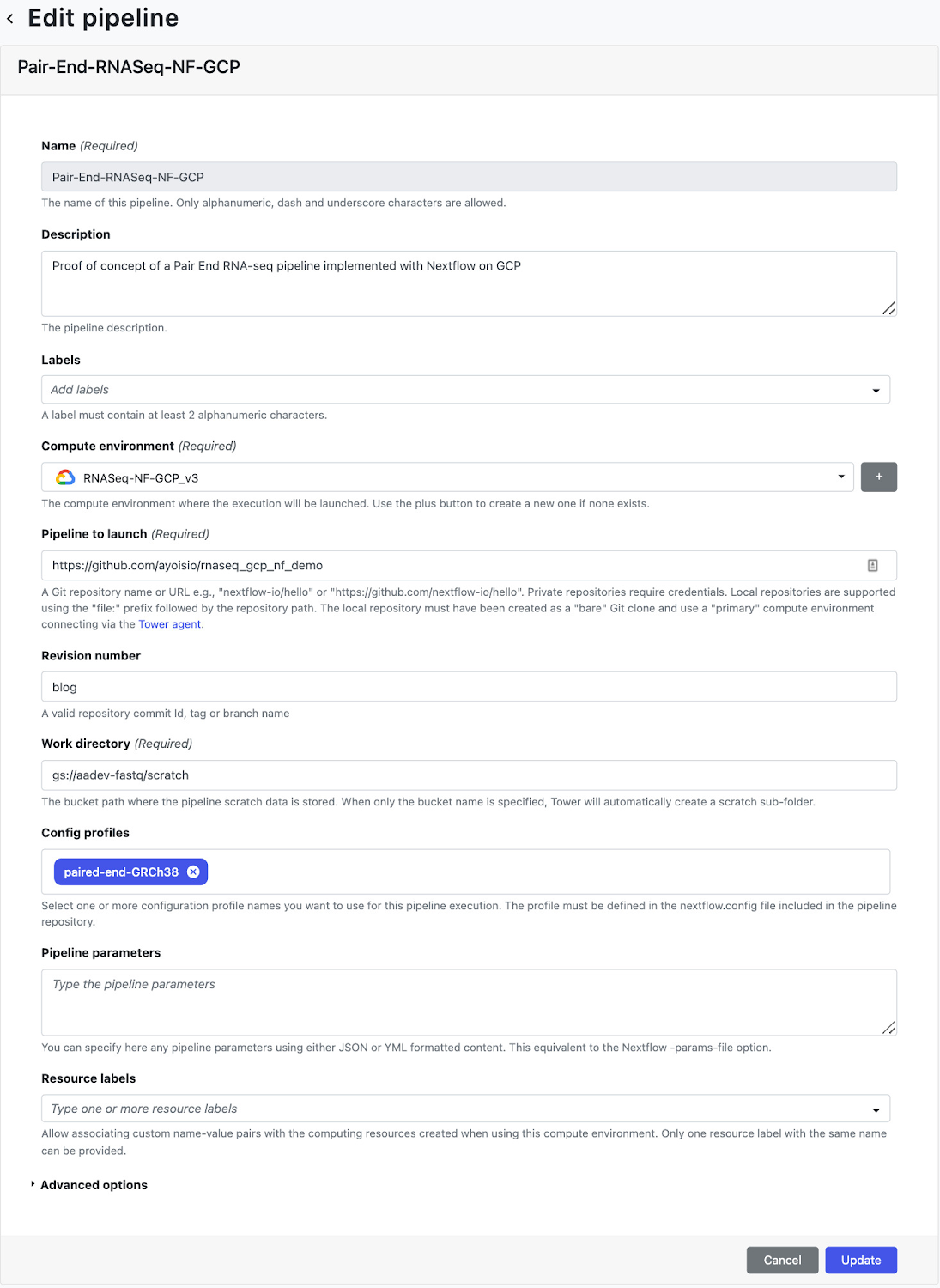
コンピューティング環境を作成したら、[Launch Pad] ページの [Add Pipeline] ビューに移動します。[Add Pipeline] ビューで、任意のパイプライン名を入力し、上記で作成したコンピューティング環境を選択します。さらに、ステップ 6 で作成した nextflow.config がホストされている Git リポジトリの URL を入力し、実行中に使用するリビジョン番号(ブランチ)を入力します。最後に、構成プロファイルとして、nextflow.config スクリプトで定義されている「paired-end-GRCh38」を入力します。
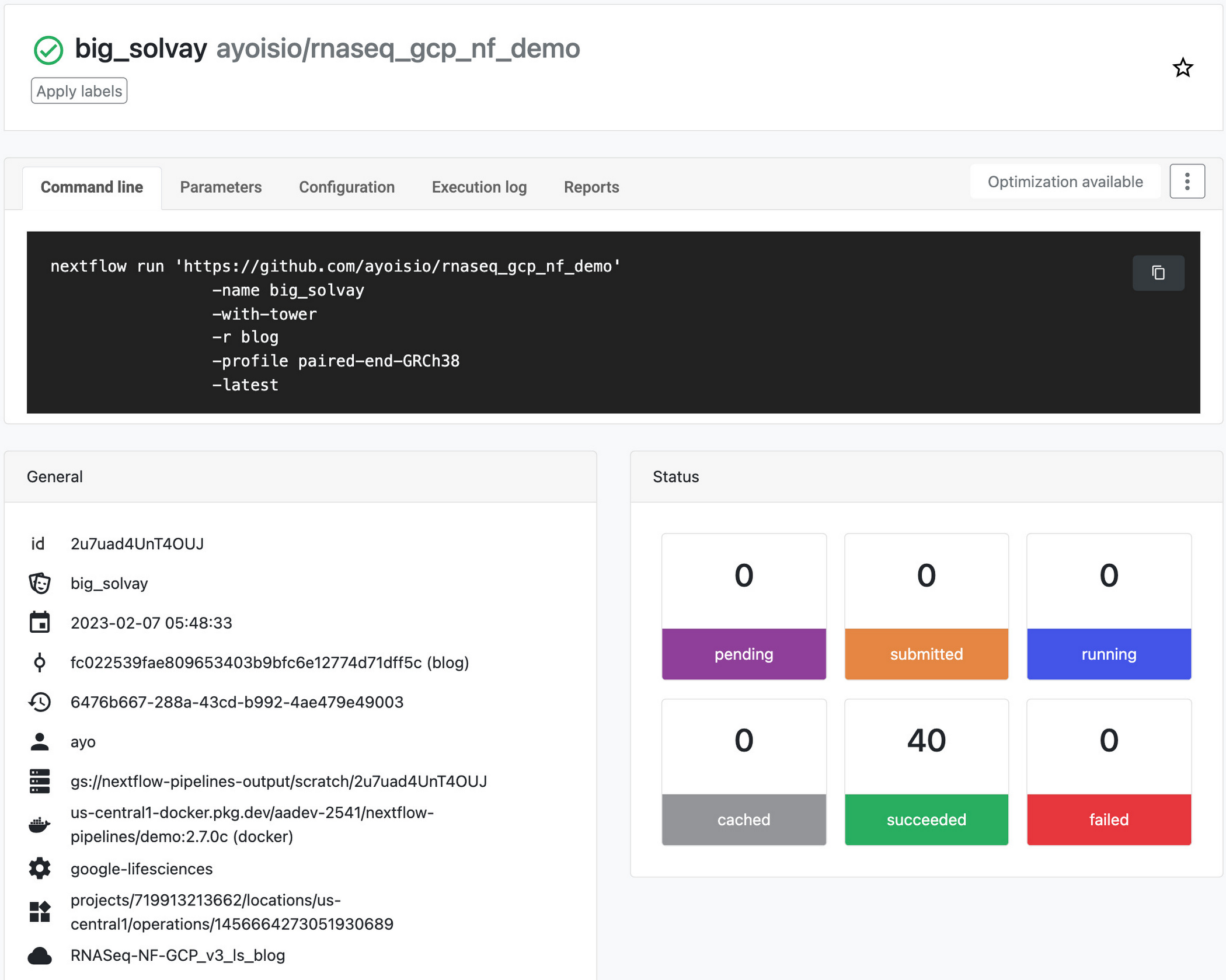
パイプラインの作成が終わったら、パイプラインを起動します。パイプラインはバックグラウンドで開始され、完了するまで 4~6 時間かかります。パイプラインの進行状況をモニタリングするには、[Runs] タブに移動してラン ID をクリックします。パイプラインが完了すると、ステップ 5 で作成したゲノムテーブルとアイソフォーム テーブルにゲノム発現とアイソフォーム発現の結果が格納され、これらのデータをクエリによって取得できます。FastQC レポートは、nextflow.config で指定した出力ディレクトリにあります。
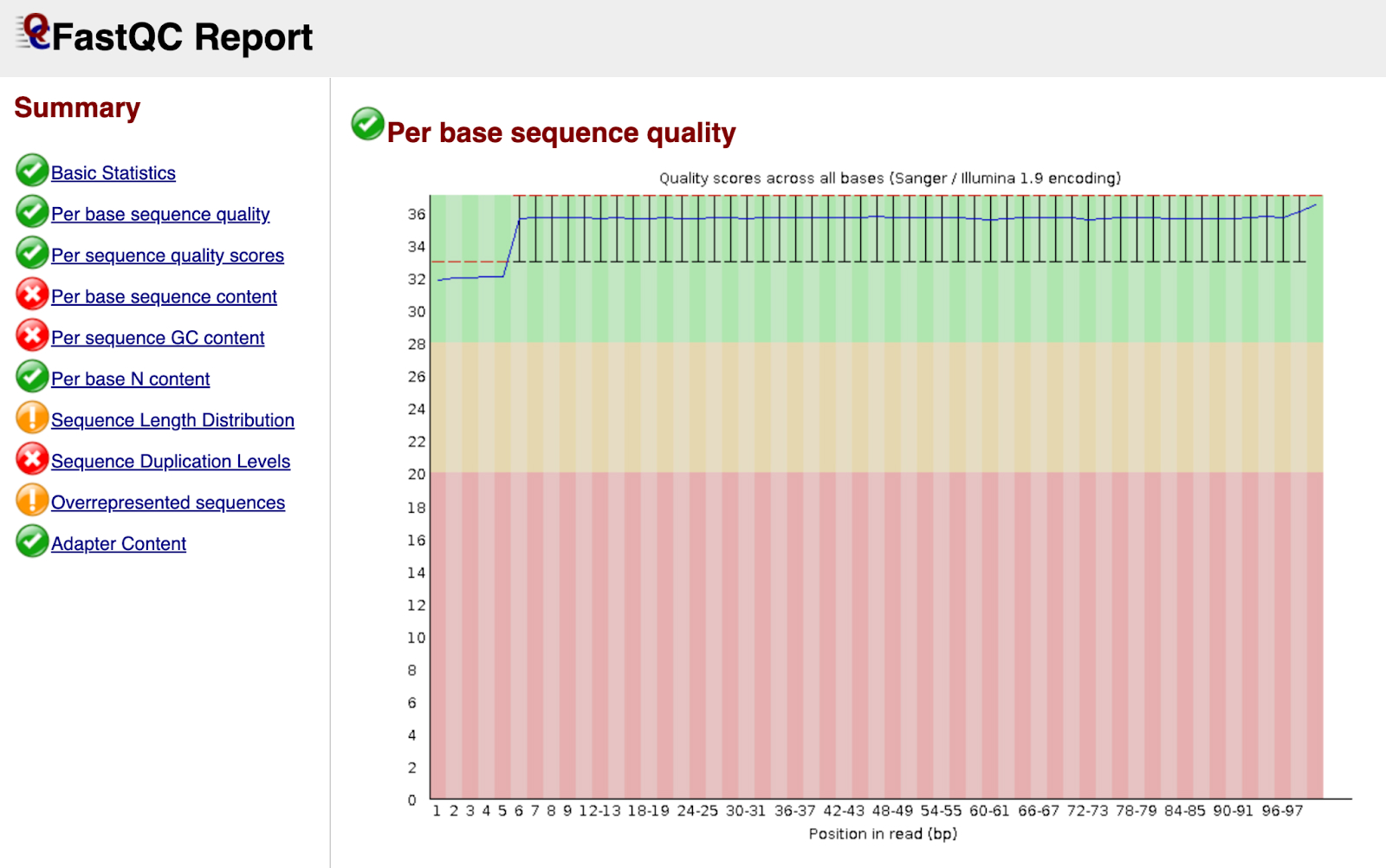
ステップ 8: DESeq2 ノートブック分析を実行する
次のステップでは、ユーザー管理の R ノートブックで、ステップ 7 で生成された BigQuery の遺伝子リード発現データを使用して DESeq2 分析を実行します。DESeq2 は、2 つ以上の生物学的状態間の差次的遺伝子発現を検出する統計メソッドを備えた R パッケージです。今回のワークフローでは、DESeq2 を使用して、ダイアモンド ブラックファン貧血の患者と対照健常者との間の差次的発現(DE)遺伝子を検出します。ここで見つかった上位の DE 遺伝子を、ステップ 10 で AlphaFold を使用して可視化します。参考用に、GitHub でノートブックが公開されています。DESeq2 分析から出力された上位 DE 遺伝子のうちのいくつか(GATA2、DYSF、TNFRSF10B など)は、ゲノム関連の文献でダイアモンド ブラックファン貧血の特徴として言及されており、ステップ 1~8 で実施した RNA シーケンス パイプライン前処理ワークフローとノートブック分析が妥当であることを証明しています。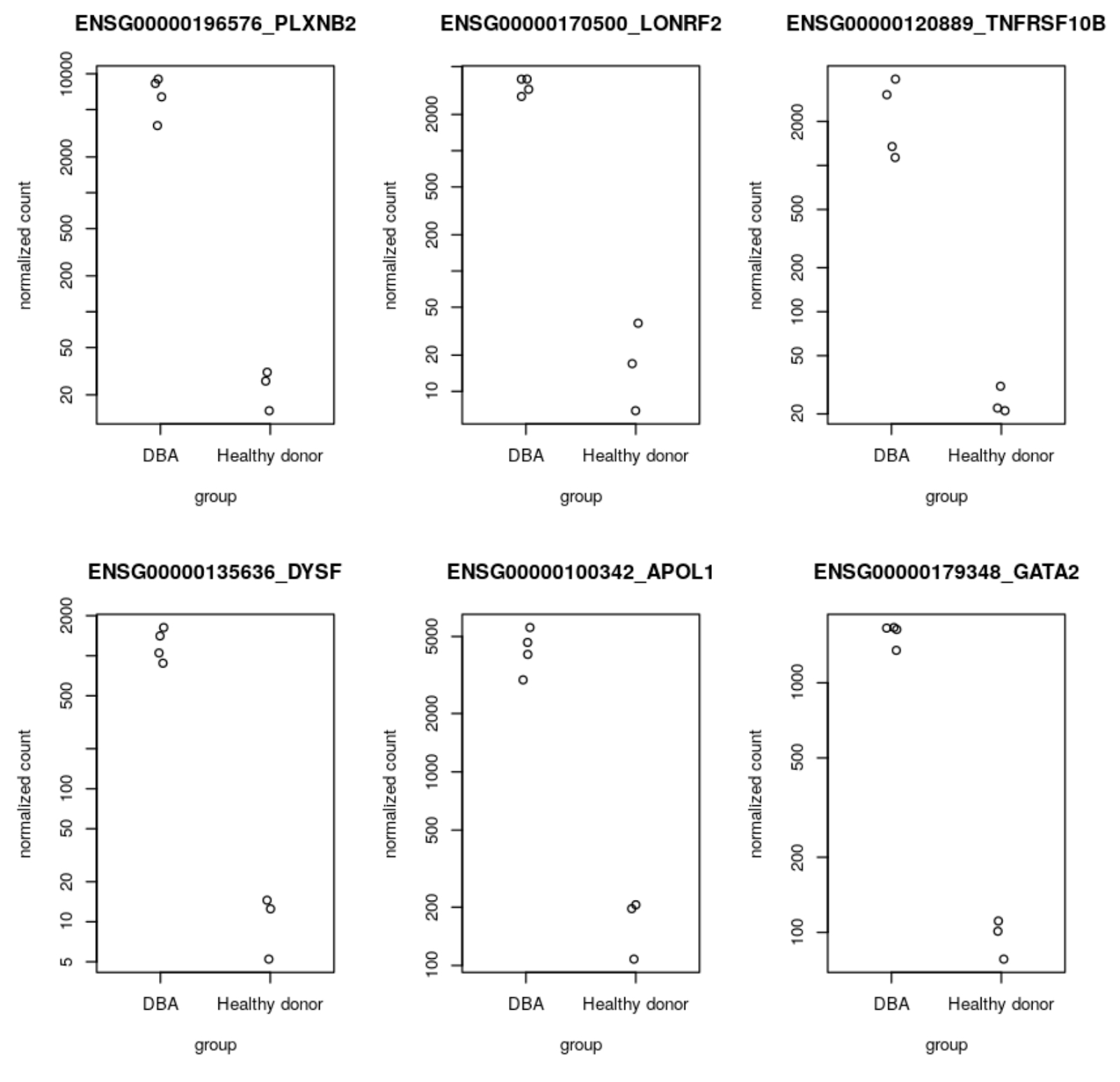
ステップ 9: 転写物をアミノ酸配列に変換するリモート関数を作成する
次のステップでは、遺伝子転写物をアミノ酸配列に変換するリモート関数を作成します。ステップ 7 で作成されたアイソフォーム発現テーブルには、遺伝子ごとのアイソフォーム発現レベルを含む転写物 ID 列が含まれます。この転写物 ID 列を、Ensembl Sequence REST API を呼び出して指定された遺伝子転写物のアミノ酸配列を出力するリモート関数に渡します。アミノ酸配列は AlphaFold への入力として使用できるため、これは RNA シーケンス パイプラインで生成された発現データから AlphaFold に直接渡すアミノ酸配列を生成するためのスケーラブルな手段となります。リモート関数の作成方法については、こちらのドキュメントをご覧ください。参考用に、GitHub でコードが公開されています。今回のワークフローでは、ステップ 8 で出力された DE 遺伝子に対応する転写物のアミノ酸配列を生成します。GATA2 転写物のアミノ酸配列を作成するサンプルクエリを以下に示します。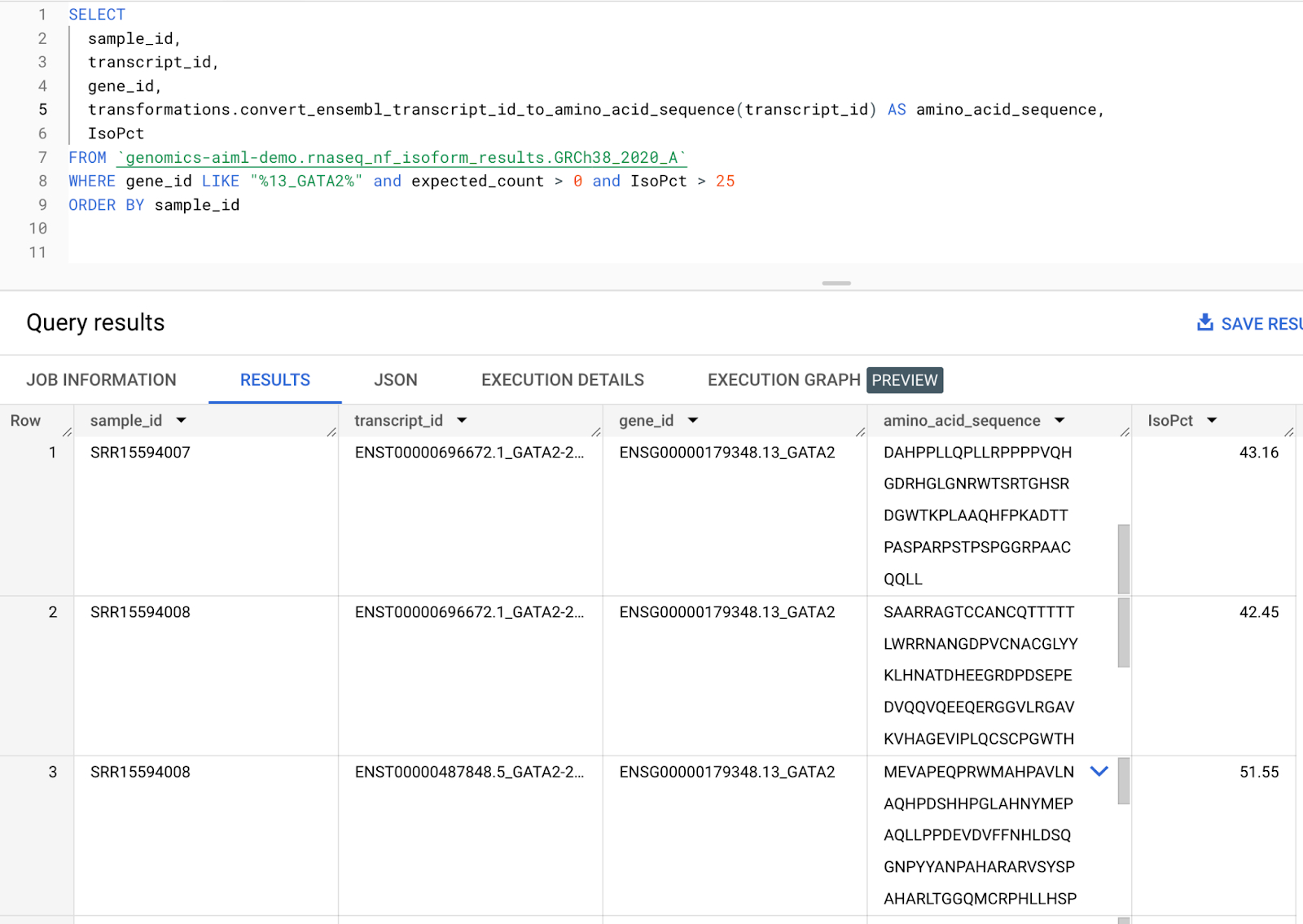
ステップ 10: AlphaFold でアミノ酸配列を可視化する
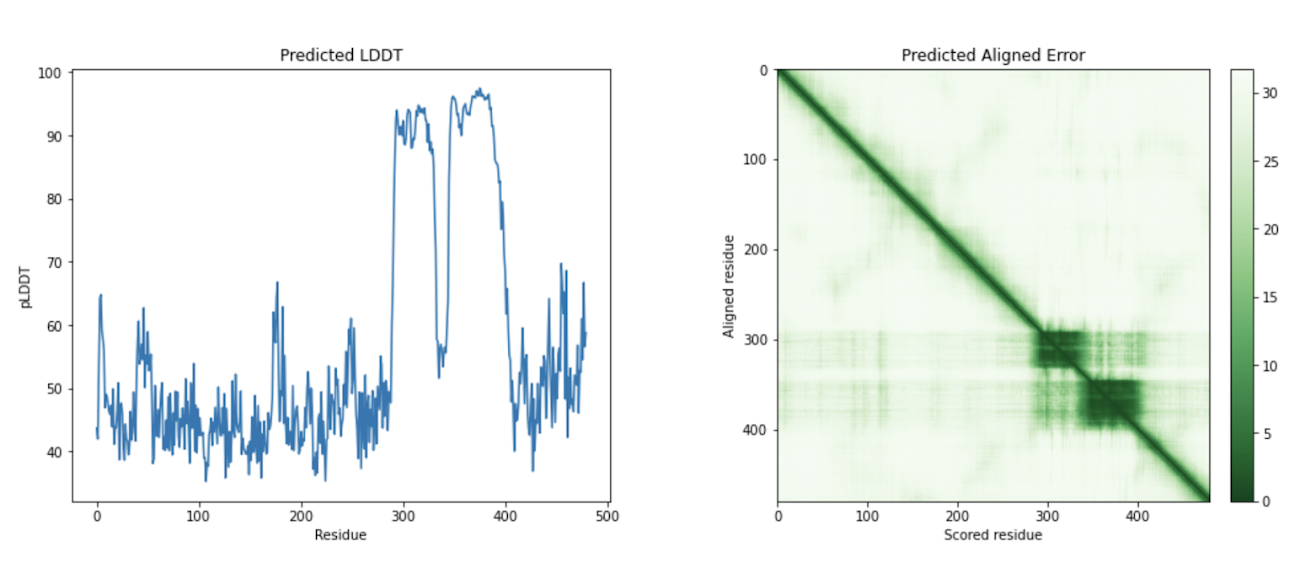
最後のステップでは、AlphaFold でアミノ酸配列を可視化します。Google Cloud では、Vertex AI Workbench でのノートブック インスタンスの起動や AlphaFold の実行に使用できるカスタマイズされた(パッケージがプリインストール済みの)Docker イメージが Artifact Registry で提供されています。ノートブック、Dockerfile、ビルド スクリプトは Vertex AI コミュニティ コンテンツにあります。推奨される GPU アクセラレータを使用してノートブックをデプロイする詳細な手順については、関連するブログ投稿をご覧ください。ここでは、BigQuery Python クライアントを使用して DE 遺伝子のアミノ酸配列を生成するクエリを実行し、それらのアミノ酸配列を AlphaFold に渡して処理します。DE 遺伝子 GATA2 のアイソフォームの一つのタンパク質構造予測の例を以下に示します。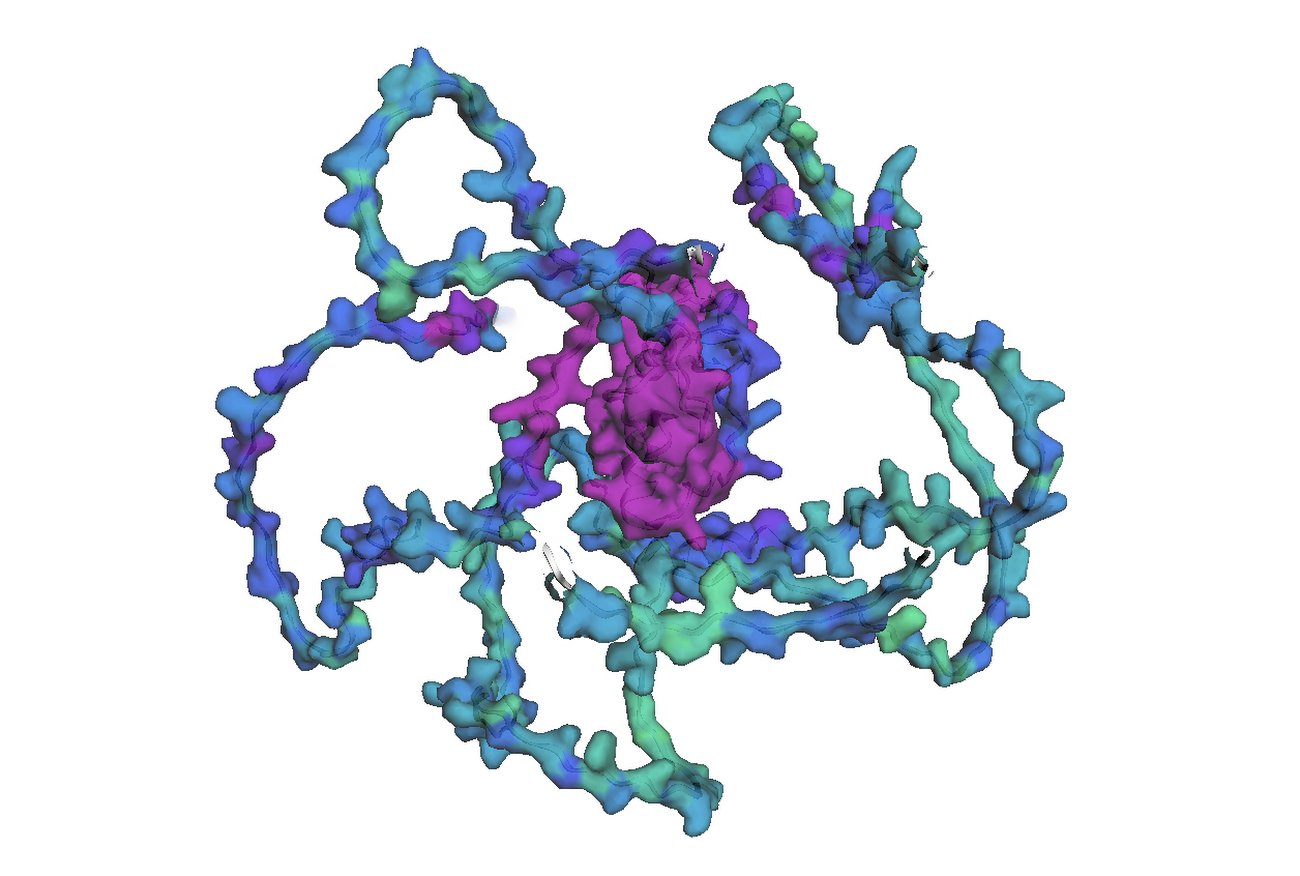
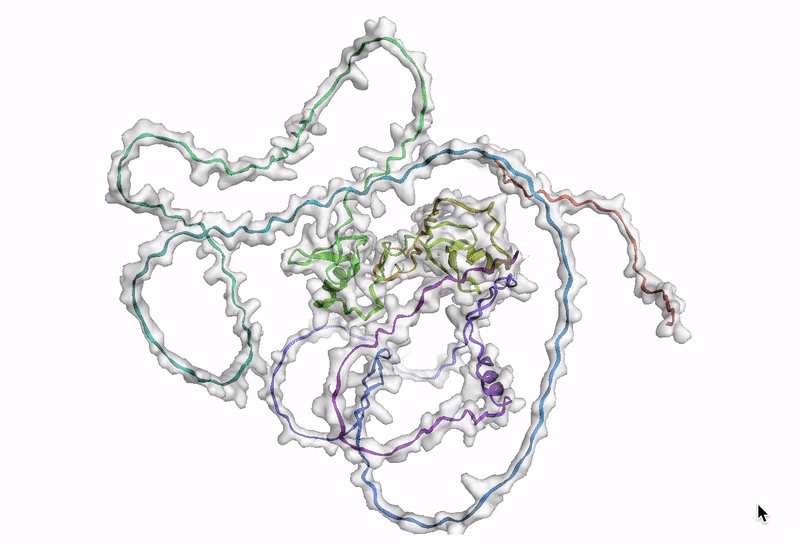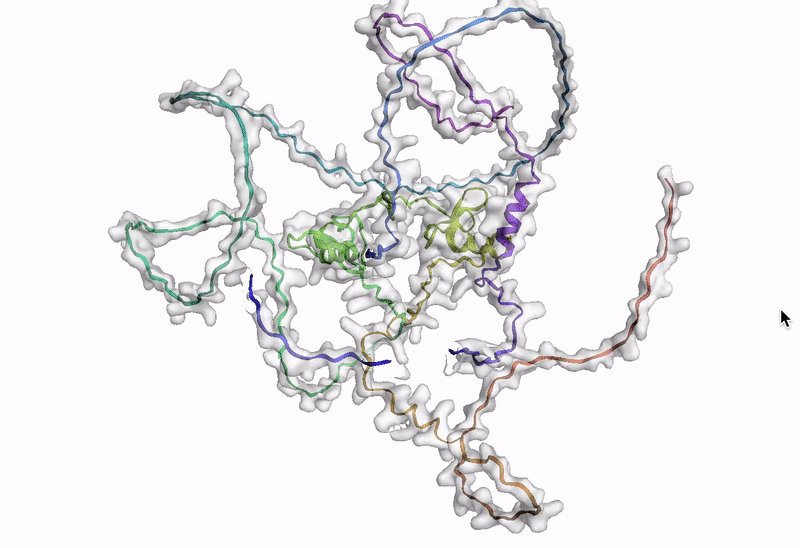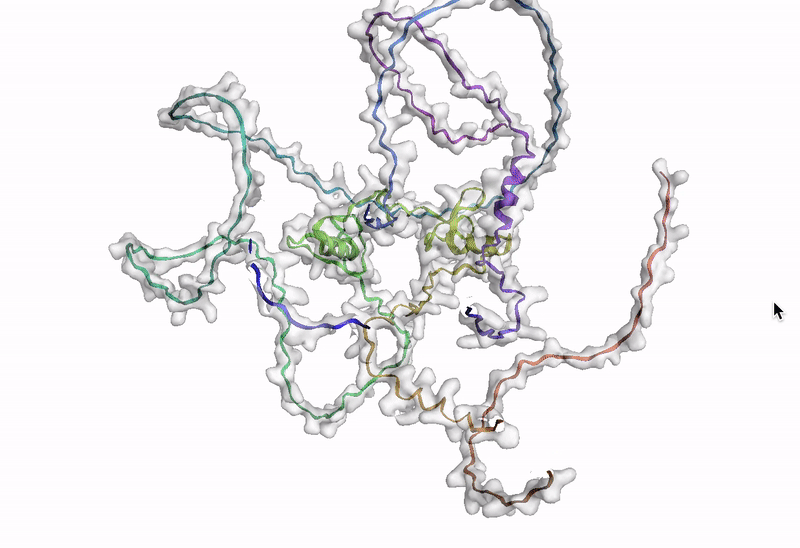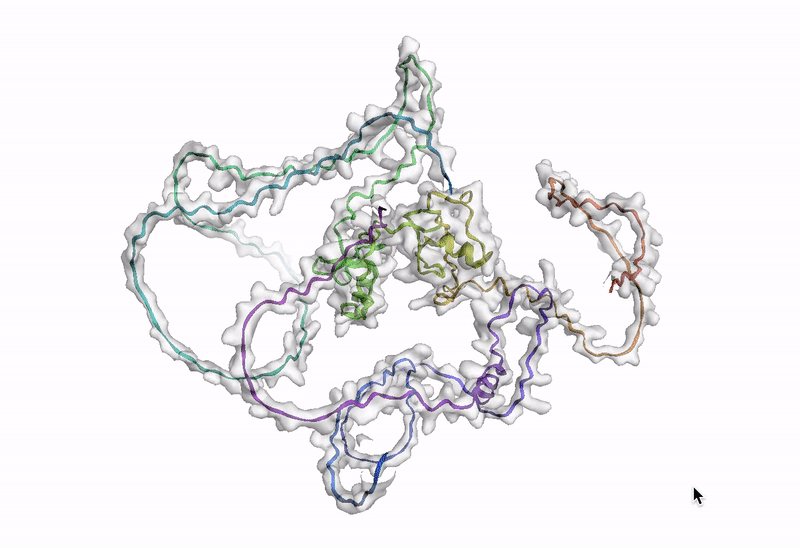
次のステップ
このワークフローを拡張し、たとえば Nextflow への並列バリアント コーリング パイプラインの実装、VCF ファイルの生成と Cloud Storage へのアップロード、Variant Transforms ツールを使用したバリアント データの BigQuery への読み込みをワークフローに追加できます。さらに、この RNA シーケンス ワークフローで得られたアイソフォーム発現データをバリアント データと組み合わせて、各サンプルに固有の、特定されたバリアントを含むアミノ酸を生成することもできます。これは、精密医療に基づくアプローチを支えるさらなる基盤となります。バリアント コーリング パイプラインの参考用に、variant-calling.nf スクリプトが GitHub で公開されています。
このブログでは、BigQuery と Vertex AI をスケーラブルに利用してゲノムデータを処理する RNA シーケンスとタンパク質構造予測のエンドツーエンドのパイプラインを開発しました。この記事が、Google Cloud を使用して現代の生物学や医学が抱えるコンピューティングの課題に立ち向かうための有益なヒントを与え、新たな発見やイノベーションへの道筋をつける助けになれば幸いです。
- HCLS、AI スペシャリスト Sarita Joshi