Genomics analysis with Hail, BigQuery, and Dataproc
Christopher Crosbie
Group Product Manager
At Google Cloud, we work with organizations performing large-scale research projects. There are a few solutions we recommend to do this type of work, so that researchers can focus on what they do best—power novel treatments, personalized medicine, and advancements in pharmaceuticals. (Find more details about creating a genomics data analysis architecture in this post.)
Hail is an open source, general-purpose, Python-based data analysis library with additional data types and methods for working with genomic data on top of Apache Spark. Hail is built to scale and has first-class support for multi-dimensional structured data, like the genomic data in a genome-wide association study (GWAS). The Hail team has made their software available to the community with the MIT license, which makes Hail a perfect augmentation to the Google Cloud Life Sciences suite of tools for processing genomics data. Dataproc makes open source data and analytics processing fast, easy, and more secure in the cloud and offers fully managed Apache Spark, which can accelerate data science with purpose-built clusters.
And what makes Google Cloud really stand out from other cloud computing platforms is our healthcare-specific tooling that makes it easy to merge genomic data with data sets from the rest of the healthcare system. When genotype data is harmonized with phenotype data from electronic health records, device data, medical notes, and medical images, the hypothesis space becomes boundless. In addition, with Google Cloud-based analysis platforms like AI Platform Notebooks and Dataproc Hub, researchers can easily work together using state-of-the-art ML tools and combine datasets in a safe and compliant manner.
Getting started with Hail and Dataproc
As of Hail version 0.2.15, pip installations of Hail come bundled with a command-line tool, hailctl, which has a submodule called dataproc for working with Dataproc clusters configured for Hail. This means getting started with Dataproc and Hail is as easy as going to the Google Cloud console, then clicking the icon for Cloud Shell at the top of the console window. This Cloud Shell provides you with command-line access to your cloud resources directly from your browser without having to install tools on your system. From this shell, you can quickly install Hail by typing:
Once Hail downloads and installs, you can create a Dataproc cluster fully configured for Apache Spark and Hail simply by running this command:
Once the Dataproc cluster is created, you can click the button from the Cloud Shell that says Open Editor, which will take you to a built-in editor for creating and modifying code. From this editor, choose New File and call the file my-first-hail-job.py.
In the editor, copy and paste the following code:
By default, Cloud Shell Editor should save this file, but you can also explicitly save the file from the menu where the file was created. Once you have verified the file is saved, return to the command line terminal by clicking on the Open Terminal button.
From the terminal, now run the command:
Once the job starts, find the Dataproc section of the Google Cloud Console and review the output of your genomics job from the Jobs tab.
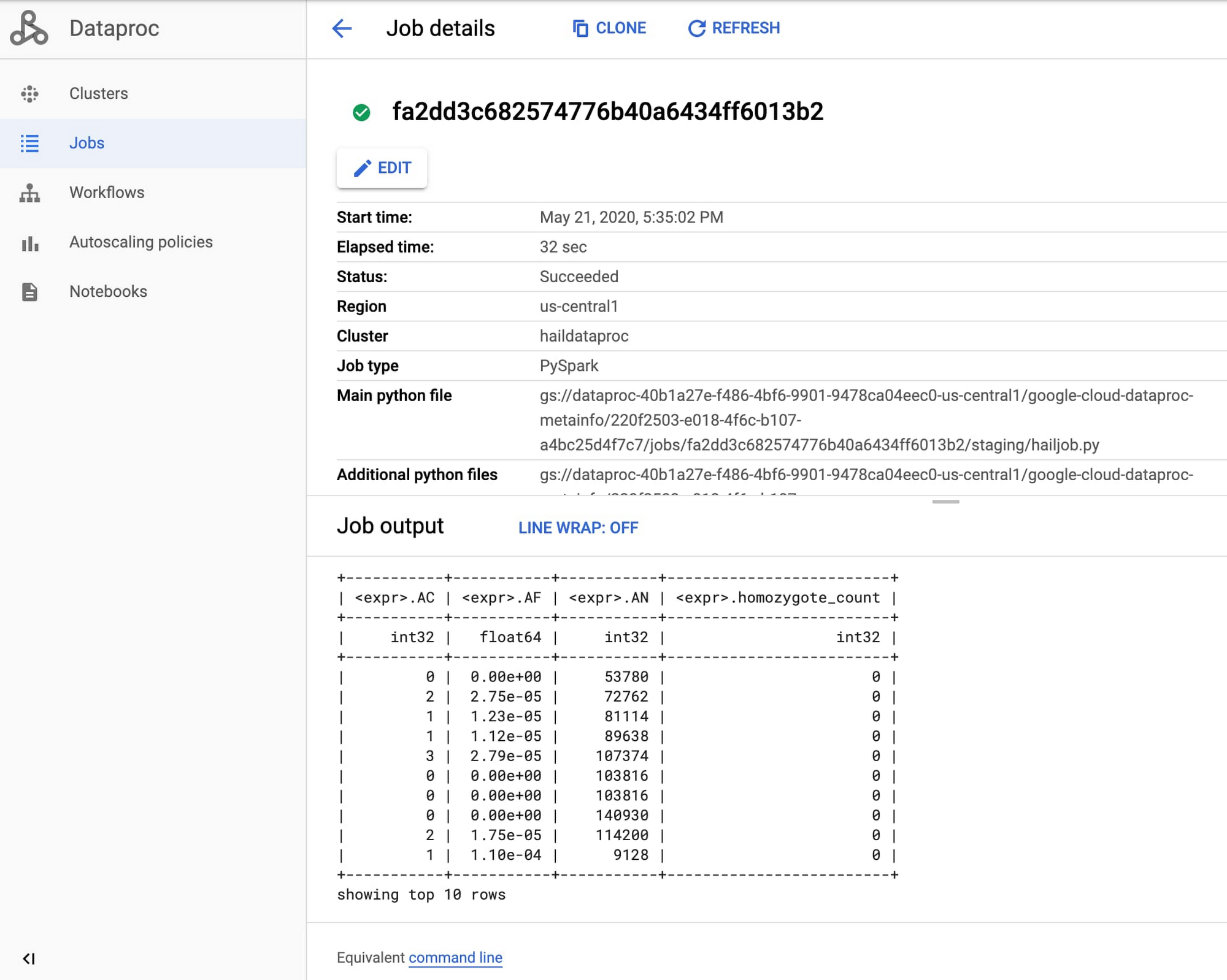
Congratulations, you just ran your first Hail job on Dataproc! For more information, see Using Hail on Google Cloud Platform.
Now, we’ll pull Dataproc and Hail into the rest of the clinical data warehouse.
Create a Dataproc Hub environment for Hail
As mentioned earlier, Hail version 0.2.15 pip installations come bundled with hailctl, a command-line tool that has a submodule called dataproc for working with Google Dataproc clusters. This includes a fully configured notebook environment that can be used simply by calling:
However, to take advantage of notebook features specific to Dataproc, including the use of Dataproc Hub, you will need to use a Dataproc initialization action that provides a standalone version of Hail without the Hail-provided notebook.
To create a Dataproc cluster that provides Hail from within Dataproc’s JuypterLab environment, run the following command:
Once the cluster has been created (as indicated by the green check mark), click on the cluster name, choose the tab for Web Interfaces and click the component gateway link for JuypterLab.
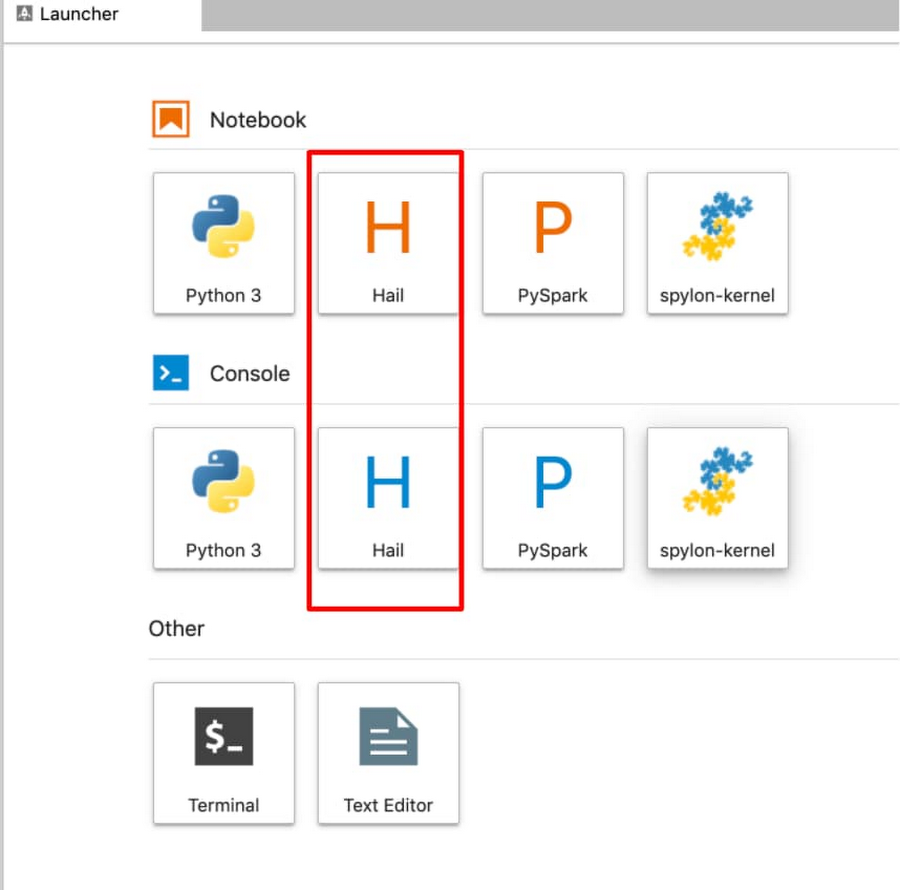
From within a Juypter IDE, you should have a kernel and console for Hail (see red box in image below):
This running cluster can easily be translated into a Dataproc Hub configuration by running the Dataproc clusters export command:
Use Dataproc Hub and BigQuery to analyze genomics data
Now that the Juypter notebooks environment is configured with Hail for Dataproc, let’s take a quick survey of the ways we can interact with the BigQuery genotype and phenotype data stored in the insights zone.
Using BigQuery magic to query data into Pandas
It is possible to run a GWAS study directly in BigQuery by using SQL logic to push the processing down into BigQuery. Then, you can bring just the query results back into a Pandas dataframe that can be visualized and presented in a notebook. From a Dataproc Juypter notebook, you can run BigQuery SQL, which returns the results in a Pandas dataframe simply by adding the bigquery magic command to the start of the notebook cell, like this:
Find an example of a GWAS analysis performed in BigQuery with a notebook in this tutorial. A feature of BigQuery, BigQuery ML provides the ability to run basic regression techniques and K-means clusters using standard SQL queries.
More commonly, BigQuery is used for preliminary steps in GWAS/PheWAS: feature engineering, defining cohorts of data, and running descriptive analysis to understand the data. Let’s look at some descriptive statistics using the 1000 genome variant data hosted by BigQuery public datasets.
Let’s say you wanted to understand what SNP data was available in chromosome 12 from the 1000 Genomes Project. In a Juypter cell, simply copy and paste the following into a cell:
This query will populate a Pandas dataframe with very basic information from the 1000 Genomes Project samples in my cohort. You can now run standard Python and Pandas functions to review, plot, and understand the data available for this cohort.
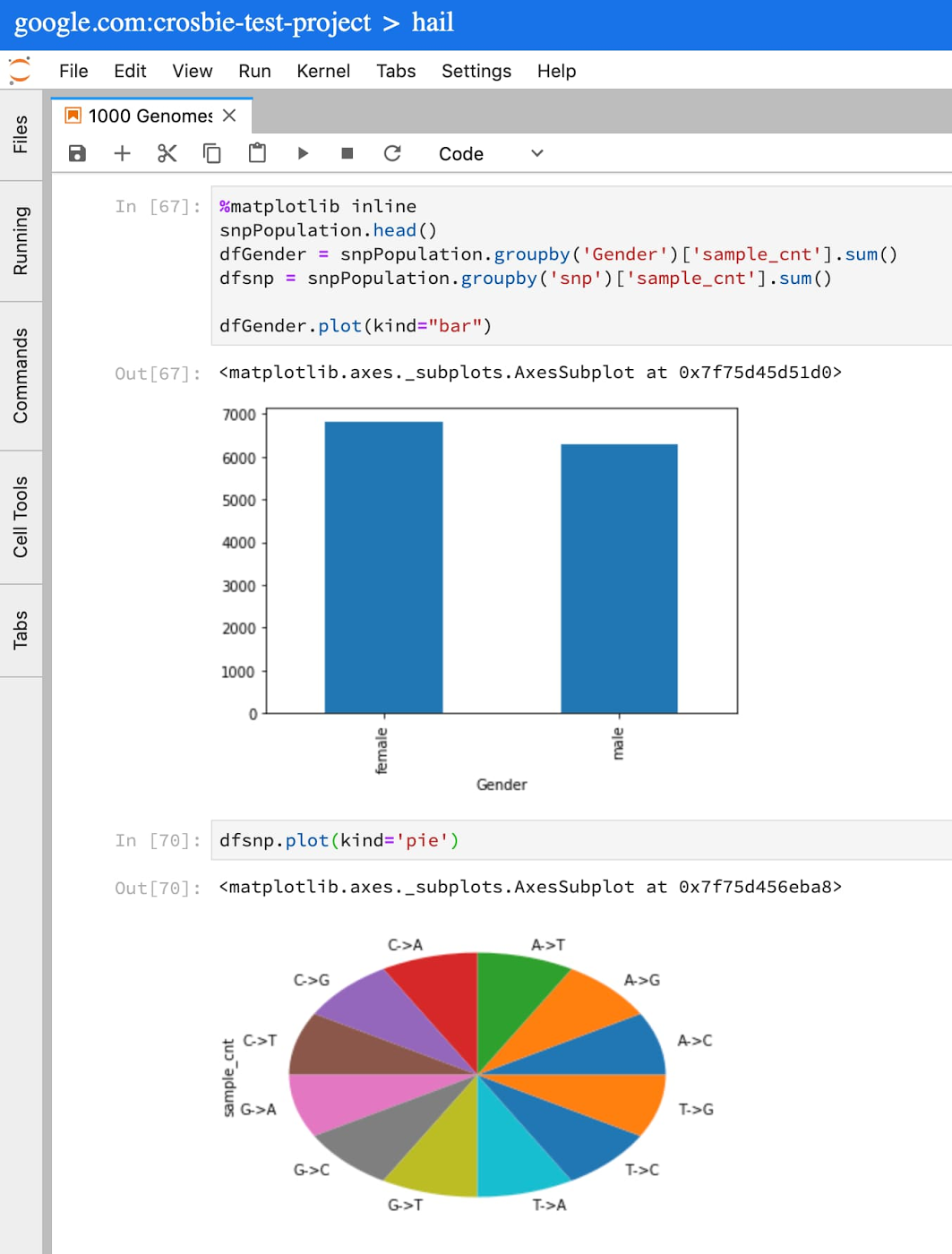
For more on writing SQL queries against tables that use the variant format, see the Advanced guide to analyzing variants using BigQuery.
Using the Spark to BigQuery connector to work with BigQuery storage directly in Apache Spark
When you need to process large volumes of genomic data for population studies and want to use generic classification and regression algorithms like Random Forest, Naive Bayes, or Gradient Boosted trees, or you need help with extracting or transforming features with algorithms like PCA or One Hot Encoding, Apache Spark offers these ML capabilities, among many others.
Using the Apache Spark BigQuery connector from Dataproc, you can now treat BigQuery as another source to read and write data from Apache Spark. This is achieved nearly the same as any other Spark dataframe setup:
Learn more here about the Apache Spark to BigQuery storage integration and how to get started.
Run variant transforms to convert BigQuery into VCF for genomics tools like Hail
When you want to do genomics-specific tasks, this is where Hail can provide a layer on top of Spark that can be used to:
Generate variant and sample annotations
Understand Mendelian violations in trios, prune variants in linkage disequilibrium, analyze genetic similarity between samples, and compute sample scores and variant loadings using PCA
Perform variant, gene-burden and eQTL association analyses using linear, logistic, and linear mixed regression, and estimate heritability
Hail expects the data format to start with either VCF, BGEN, or PLINK. Luckily, BigQuery genomics data can easily be converted from the BigQuery VCF format into a VCF file using Variant Transforms. Once you create the VCF on Cloud Storage, call Hail’s import_vcf function, which transforms the file into Hail’s matrix table.
To learn more about scalable genomics analysis with Hail, check out this YouTube series for Hail from the Broad Institute.



