Questa procedura dettagliata illustra come pre-elaborare, addestrare ed eseguire previsioni su un modello di machine learning utilizzando Apache Beam, Google Dataflow e TensorFlow.
Per dimostrare questi concetti, questa procedura dettagliata utilizza l'esempio di codice Molecules. Dati i dati molecolari come input, l'esempio di codice Molecules crea e addestra un modello di machine learning per prevedere l'energia molecolare.
Costi
Questa procedura dettagliata utilizza potenzialmente componenti fatturabili di Google Cloud, inclusi uno o più dei seguenti elementi:
- Dataflow
- Cloud Storage
- AI Platform
Utilizza il Calcolatore prezzi per generare una stima dei costi in base all'utilizzo previsto.
Panoramica
L'esempio di codice Molecules estrae file che contengono dati molecolari e conta il numero di atomi di carbonio, idrogeno, ossigeno e azoto presenti in ogni molecola. Quindi, il codice normalizza i conteggi a valori compresi tra 0 e 1 e li inserisce in uno strumento di stima TensorFlow Deep Neural Network. Lo strumento per la stima della rete neurale addestra un modello di machine learning per prevedere l'energia molecolare.
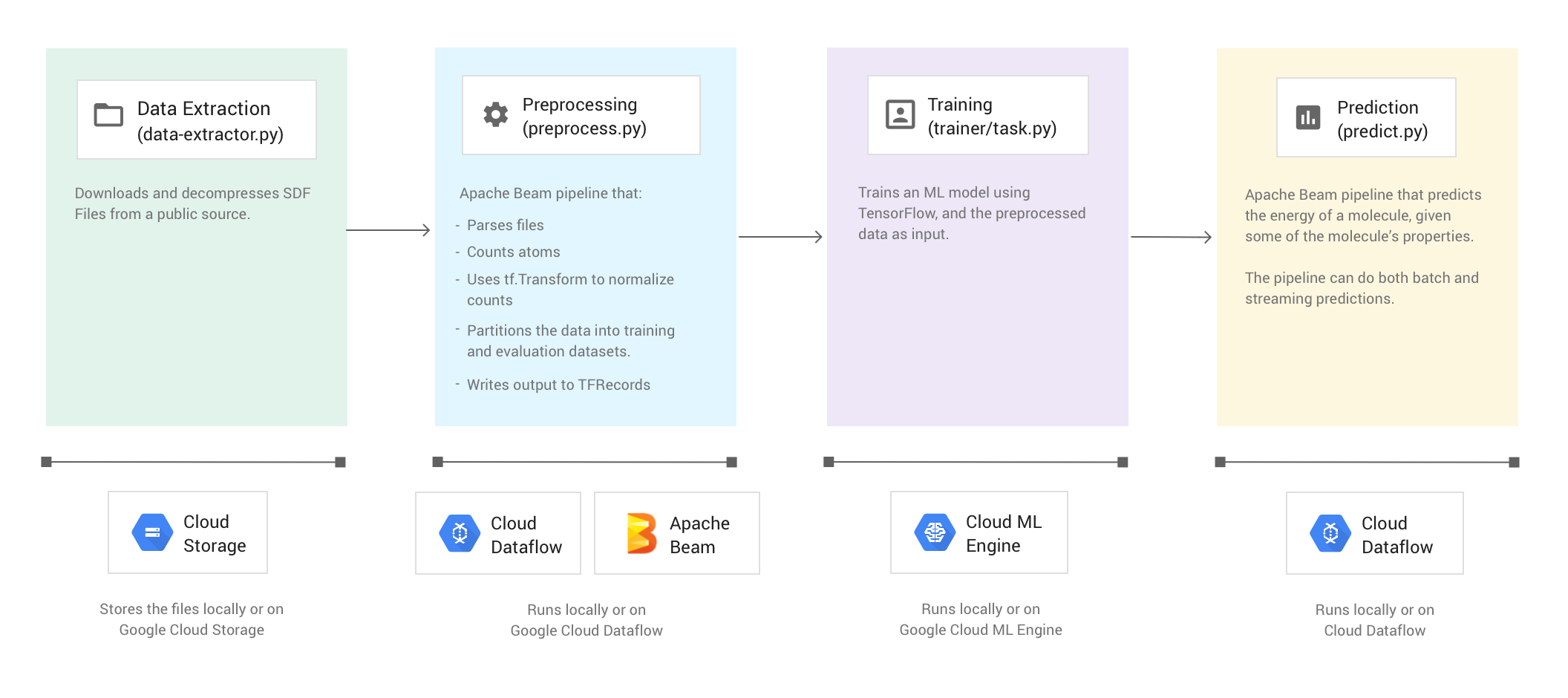
L'esempio di codice si compone di quattro fasi:
- Estrazione dei dati (
data-extractor.py) - Pre-elaborazione (
preprocess.py) - Addestramento (
trainer/task.py) - Previsione (
predict.py)
Le sezioni che seguono affrontano le quattro fasi, ma questa procedura dettagliata si concentra maggiormente sulle fasi che utilizzano Apache Beam e Dataflow: la fase di pre-elaborazione e la fase di previsione. La fase di pre-elaborazione utilizza anche
la libreria Transform TensorFlow (comunemente nota come tf.Transform).
L'immagine seguente mostra il flusso di lavoro dell'esempio di codice Molecules.
Esegui l'esempio di codice
Per configurare l'ambiente, segui le istruzioni nel README del repository GitHub di Molecules. Quindi, esegui l'esempio di codice Molecules utilizzando uno degli script wrapper forniti, run-local o run-cloud. Questi script eseguono automaticamente tutte e quattro le fasi dell'esempio di codice (estrazione, pre-elaborazione, addestramento e previsione).
In alternativa, puoi eseguire ogni fase manualmente utilizzando i comandi forniti nelle sezioni di questo documento.
Esegui localmente
Per eseguire localmente l'esempio di codice Molecules, esegui lo script del wrapper run-local:
./run-local
I log di output mostrano quando lo script esegue ciascuna delle quattro fasi (estrazione dei dati, pre-elaborazione, addestramento e previsione).
Lo script data-extractor.py contiene un argomento obbligatorio per il numero di file.
Per motivi di facilità d'uso, lo script run-local e gli script run-cloud hanno un valore predefinito di 5 file per questo argomento. Ogni file contiene 25.000 molecole. L'esecuzione dell'esempio di codice dovrebbe richiedere circa 3-7 minuti dall'inizio alla fine. Il tempo necessario per l'esecuzione varia in base alla CPU del computer.
Esegui su Google Cloud
Per eseguire l'esempio di codice Molecules su Google Cloud, esegui lo script wrapper run-cloud. Tutti i file di input devono essere in Cloud Storage.
Imposta il parametro --work-dir sul bucket Cloud Storage:
./run-cloud --work-dir gs://<your-bucket-name>/cloudml-samples/molecules
Fase 1: estrazione dei dati
Codice sorgente: data-extractor.py
Il primo passaggio consiste nell'estrazione dei dati di input. Il file data-extractor.py estrae e decomprime i file SDF specificati. Nei passaggi successivi, l'esempio preelabora questi file e utilizza i dati per addestrare e valutare il modello di machine learning. Il file estrae i file SDF dalla fonte pubblica e li archivia in una sottodirectory all'interno della directory di lavoro specificata. La directory di lavoro predefinita (--work-dir) è /tmp/cloudml-samples/molecules.
Archivia i file estratti
Archivia i file di dati estratti localmente:
python data-extractor.py --max-data-files 5
In alternativa, archivia i file di dati estratti in una posizione di Cloud Storage:
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python data-extractor.py --work-dir $WORK_DIR --max-data-files 5
Fase 2. Pre-elaborazione
Codice sorgente: preprocess.py
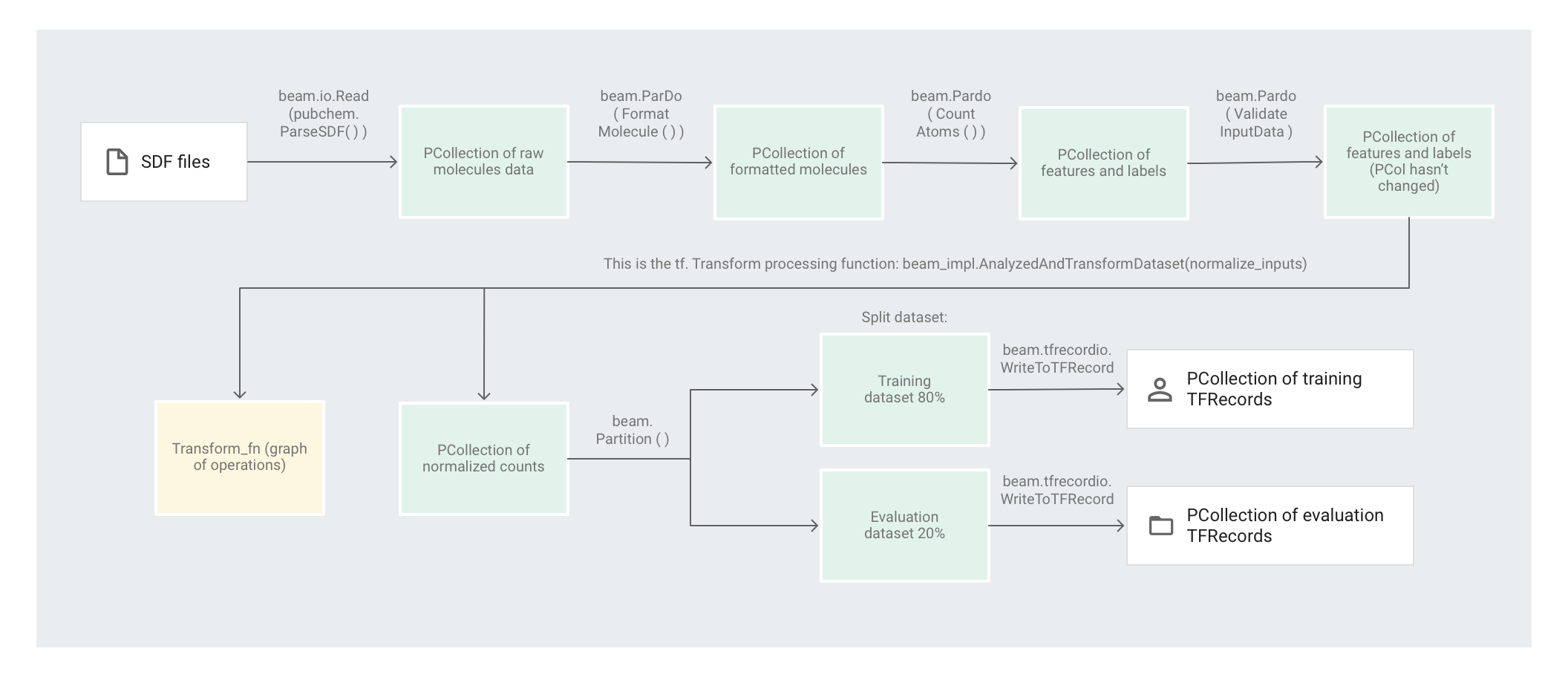
L'esempio di codice Molecules utilizza una pipeline Apache Beam per pre-elaborare i dati. La pipeline esegue le seguenti azioni di pre-elaborazione:
- Legge e analizza i file SDF estratti.
- Conta il numero di diversi atomi in ciascuna delle molecole dei file.
- Normalizza i conteggi a valori compresi tra 0 e 1 utilizzando
tf.Transform. - Partiziona il set di dati in un set di dati di addestramento e in un set di dati di valutazione.
- Scrive i due set di dati come oggetti
TFRecord.
Le trasformazioni di Apache Beam possono manipolare in modo efficiente singoli elementi alla volta, ma le trasformazioni che richiedono il passaggio completo del set di dati non possono essere eseguite facilmente solo con Apache Beam, ma utilizzando al meglio tf.Transform. Per questo motivo, il codice utilizza le trasformazioni di Apache Beam per leggere e formattare le molecole, nonché per conteggiare gli atomi di ciascuna molecola. Il codice utilizza quindi tf.Transform per trovare i conteggi minimi e massimi globali per normalizzare i dati.
L'immagine seguente mostra i passaggi nella pipeline.
Applicazione delle trasformazioni basate su elementi
Il codice preprocess.py crea una pipeline Apache Beam.
Successivamente, il codice applica una trasformazione feature_extraction alla pipeline.
La pipeline utilizza SimpleFeatureExtraction come sua trasformazione feature_extraction.
La trasformazione SimpleFeatureExtraction, definita in pubchem/pipeline.py, contiene una serie di trasformazioni che manipolano tutti gli elementi in modo indipendente.
In primo luogo, il codice analizza le molecole presenti nel file sorgente, quindi le formatta
in un dizionario di proprietà delle molecole e infine conta gli atomi
nella molecola. Questi conteggi sono le funzionalità (input) per il modello di machine learning.
La trasformazione di lettura beam.io.Read(pubchem.ParseSDF(data_files_pattern)) legge i file SDF da un'origine personalizzata.
L'origine personalizzata, chiamata ParseSDF, è definita in pubchem/pipeline.py.
ParseSDF estende FileBasedSource
e implementa la funzione read_records che apre i file SDF estratti.
Quando esegui l'esempio di codice Molecules su Google Cloud, più lavoratori (VM) possono leggere i file contemporaneamente. Per garantire che due worker non leggano gli stessi contenuti nei file, ogni file utilizza un elemento range_tracker.
La pipeline raggruppa i dati non elaborati in sezioni di informazioni pertinenti necessarie per i passaggi successivi. Ogni sezione nel file SDF analizzato viene archiviata in un dizionario (vedi pipeline/sdf.py), dove le chiavi sono i nomi delle sezioni e i valori sono i contenuti delle righe non elaborate della sezione corrispondente.
Il codice applica beam.ParDo(FormatMolecule()) alla pipeline. ParDo applica il valore DoFn denominato FormatMolecule a ciascuna molecola. FormatMolecule restituisce un dizionario di molecole formattate. Il seguente snippet è un esempio di un elemento nella PCollection di output:
{
'atoms': [
{
'atom_atom_mapping_number': 0,
'atom_stereo_parity': 0,
'atom_symbol': u'O',
'charge': 0,
'exact_change_flag': 0,
'h0_designator': 0,
'hydrogen_count': 0,
'inversion_retention': 0,
'mass_difference': 0,
'stereo_care_box': 0,
'valence': 0,
'x': -0.0782,
'y': -1.5651,
'z': 1.3894,
},
...
],
'bonds': [
{
'bond_stereo': 0,
'bond_topology': 0,
'bond_type': 1,
'first_atom_number': 1,
'reacting_center_status': 0,
'second_atom_number': 5,
},
...
],
'<PUBCHEM_COMPOUND_CID>': ['3\n'],
...
'<PUBCHEM_MMFF94_ENERGY>': ['19.4085\n'],
...
}
Quindi il codice applica beam.ParDo(CountAtoms()) alla pipeline. Il valore DoFn
CountAtoms somma il numero di atomi di carbonio, idrogeno, azoto e ossigeno
che ogni molecola ha. CountAtoms genera una PCollection di funzionalità ed etichette.
Ecco un esempio di un elemento nella PCollection di output:
{
'ID': 3,
'TotalC': 7,
'TotalH': 8,
'TotalO': 4,
'TotalN': 0,
'Energy': 19.4085,
}
La pipeline convalida quindi gli input. L'elemento ValidateInputData DoFn convalida
che ogni elemento corrisponda ai metadati specificati in input_schema. Questa convalida garantisce che i dati siano nel formato corretto quando vengono inviati a TensorFlow.
Applicazione delle trasformazioni del pass completo
L'esempio di codice Molecules utilizza un Deep Neural Network Regressor per fare previsioni. Il consiglio generale è di normalizzare gli input prima di inserirli nel modello ML. La pipeline utilizza tf.Transform per normalizzare i conteggi di ogni atomo a valori compresi tra 0 e 1. Per scoprire di più sulla normalizzazione degli input, consulta Scalabilità delle funzionalità.
La normalizzazione dei valori richiede un passaggio completo del set di dati, registrando i valori minimo e massimo. Il codice utilizza tf.Transform per esaminare l'intero set di dati e applicare le trasformazioni full-pass.
Per utilizzare tf.Transform, il codice deve fornire una funzione contenente la logica
della trasformazione da eseguire sul set di dati. In preprocess.py, il codice utilizza la trasformazione AnalyzeAndTransformDataset fornita da tf.Transform. Scopri di più su
come utilizzare tf.Transform.
In preprocess.py, la funzione feature_scaling utilizzata è normalize_inputs,
definita in pubchem/pipeline.py. La funzione utilizza la funzione tf.Transform scale_to_0_1 per normalizzare i conteggi sui valori compresi tra 0 e 1.
È possibile normalizzare i dati manualmente, ma se il set di dati è di grandi dimensioni è più veloce utilizzare Dataflow. L'uso di Dataflow consente di eseguire la pipeline su più worker (VM) in base alle necessità.
Partizionamento del set di dati
Successivamente, la pipeline preprocess.py partiziona il singolo set di dati in due set di dati. Alloca circa l'80% dei dati da utilizzare come dati di addestramento e circa il 20% da utilizzare come dati di valutazione.
Scrittura dell'output
Infine, la pipeline preprocess.py scrive i due set di dati (addestramento e valutazione) utilizzando la trasformazione WriteToTFRecord.
Esegui la pipeline di pre-elaborazione
Esegui la pipeline di pre-elaborazione in locale:
python preprocess.py
In alternativa, esegui la pipeline di pre-elaborazione su Dataflow:
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python preprocess.py \
--project $PROJECT \
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--work-dir $WORK_DIR
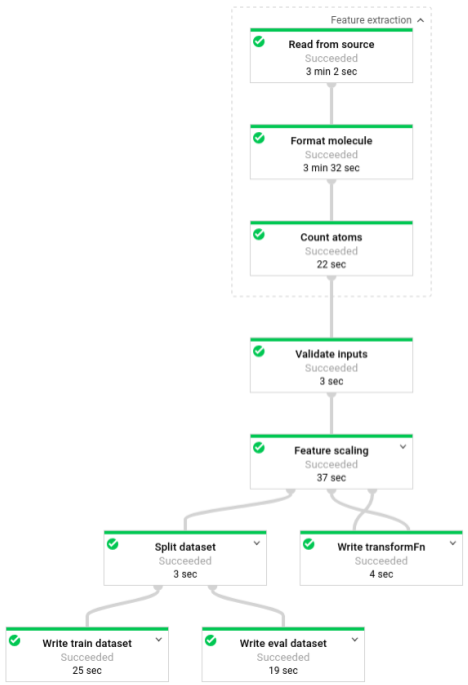
Quando la pipeline è in esecuzione, puoi visualizzarne l'avanzamento nell'interfaccia di monitoraggio di Dataflow:
Fase 3: addestramento
Codice sorgente: trainer/task.py
Ricorda che, al termine della fase di pre-elaborazione, il codice suddivide i dati in due set di dati (addestramento e valutazione).
L'esempio utilizza TensorFlow per addestrare il modello di machine learning. Il file trainer/task.py nell'esempio di codice Molecules contiene il codice per addestrare il modello. La funzione principale di trainer/task.py carica i dati elaborati nella fase di pre-elaborazione.
Lo strumento di stima utilizza il set di dati di addestramento per addestrare il modello, quindi utilizza il set di dati di valutazione per verificare che il modello preveda con precisione l'energia molecolare in base ad alcune proprietà della molecola.
Scopri di più sull'addestramento di un modello di ML.
Addestra il modello
Addestra il modello in locale:
python trainer/task.py
# To get the path of the trained model
EXPORT_DIR=/tmp/cloudml-samples/molecules/model/export/final
MODEL_DIR=$(ls -d -1 $EXPORT_DIR/* | sort -r | head -n 1)
In alternativa, addestra il modello su AI Platform:
- Seleziona prima una regione supportata per eseguire il job di addestramento
gcloud config set compute/region $REGION
- Avviare il job di addestramento
JOB="cloudml_samples_molecules_$(date +%Y%m%d_%H%M%S)"
BUCKET=gs://<your bucket name>
WORK_DIR=$BUCKET/cloudml-samples/molecules
gcloud ai-platform jobs submit training $JOB \
--module-name trainer.task \
--package-path trainer \
--staging-bucket $BUCKET \
--runtime-version 1.13 \
--stream-logs \
-- \
--work-dir $WORK_DIR
# To get the path of the trained model:
EXPORT_DIR=$WORK_DIR/model/export
MODEL_DIR=$(gsutil ls -d $EXPORT_DIR/* | sort -r | head -n 1)
Fase 4. Previsione
Codice sorgente: predict.py
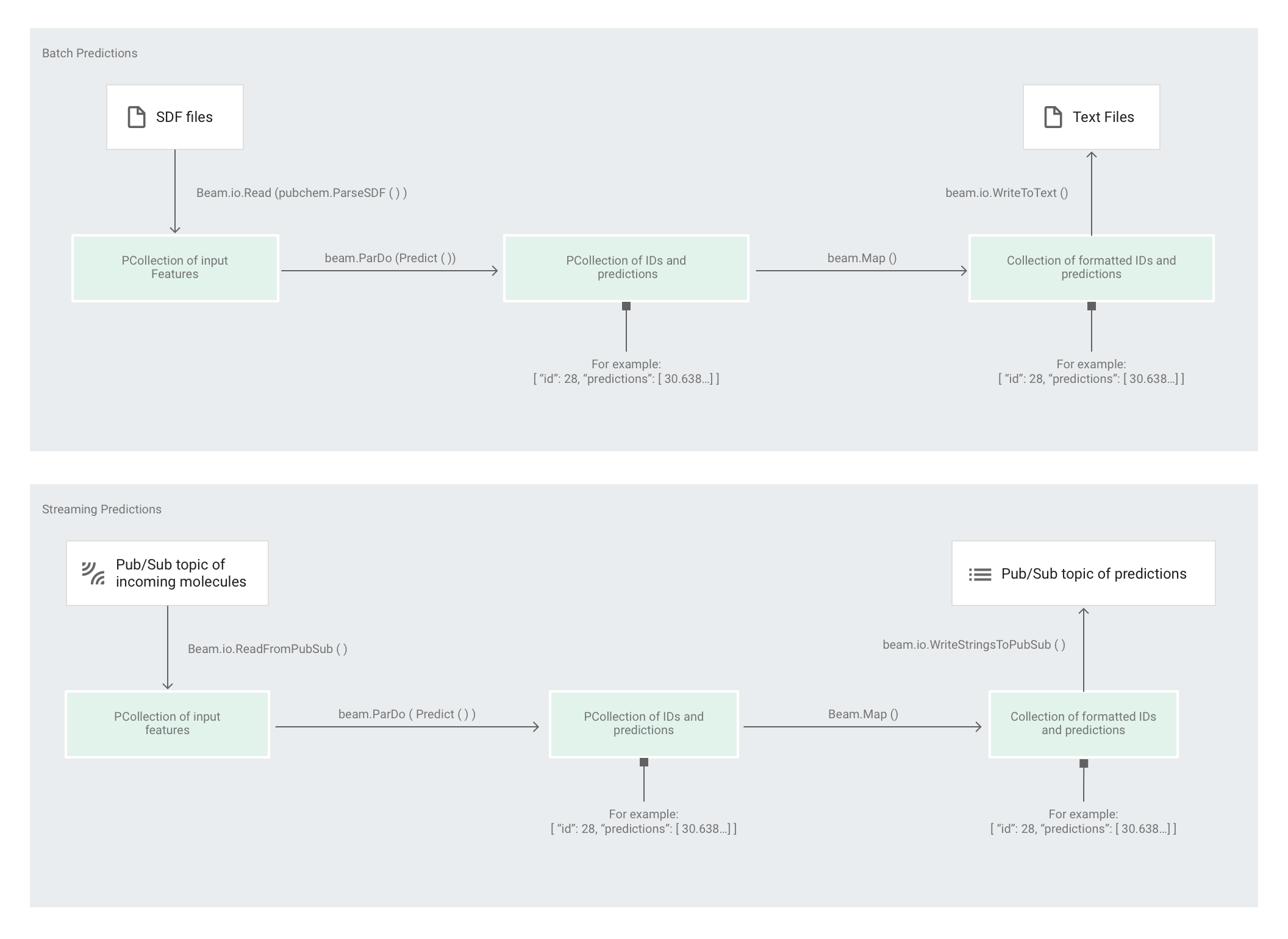
Dopo che lo strumento di stima ha addestrato il modello, puoi fornirgli input e fare previsioni. Nell'esempio di codice Molecules, la pipeline in predict.py è responsabile dell'esecuzione delle previsioni. La pipeline può fungere da pipeline in modalità batch o pipeline in modalità flusso.
Il codice per la pipeline è lo stesso per batch e streaming, ad eccezione delle interazioni di origine e sink. Se la pipeline viene eseguita in modalità batch, legge i file di input dalla origine personalizzata e scrive le previsioni di output come file di testo nella directory di lavoro specificata. Se la pipeline viene eseguita in modalità flusso, legge le molecole di input quando arrivano da un argomento Pub/Sub. La pipeline scrive le previsioni di output, quando sono pronte, in un argomento Pub/Sub diverso.
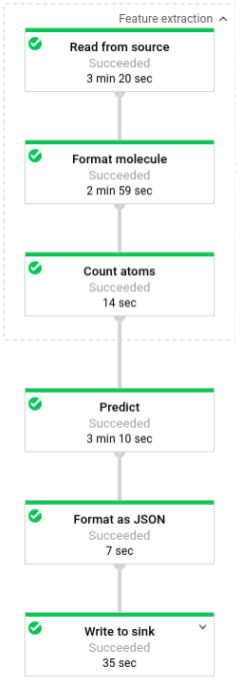
L'immagine seguente mostra i passaggi nelle pipeline di previsione (batch e flusso).
In predict.py, il codice definisce la pipeline nella funzione run:
Il codice chiama la funzione run con i seguenti parametri:
Innanzitutto, il codice trasmette la trasformazione pubchem.SimpleFeatureExtraction(source) durante la trasformazione feature_extraction. Questa trasformazione, utilizzata anche nella fase di pre-elaborazione, viene applicata alla pipeline:
La trasformazione trasforma la lettura dalla sorgente appropriata in base alla modalità di esecuzione della pipeline (batch o flusso), formatta le molecole e conta i diversi atomi di ciascuna molecola.
Quindi, beam.ParDo(Predict(…)) viene applicato alla pipeline che esegue la
previsione dell'energia molecolare.
Predict, l'DoFn passato, utilizza il dizionario specifico delle funzionalità di input
(conteggio atomico) per prevedere l'energia molecolare.
La trasformazione successiva applicata alla pipeline è
beam.Map(lambda result: json.dumps(result)), che prende il dizionario dei risultati della previsione
e lo serializza in una stringa JSON.
Infine, l'output viene scritto nel sink (sia come file di testo nella directory di lavoro per la modalità batch sia come messaggi pubblicati in un argomento Pub/Sub per la modalità di streaming).
Previsioni batch
Le previsioni batch sono ottimizzate per la velocità effettiva anziché per la latenza. Le previsioni batch funzionano meglio se esegui molte previsioni e puoi attendere che tutte le previsioni vengano completate prima di ottenere i risultati.
Esegui la pipeline di previsione in modalità batch
Esegui la pipeline di previsione batch in locale:
# For simplicity, we'll use the same files we used for training
python predict.py \
--model-dir $MODEL_DIR \
batch \
--inputs-dir /tmp/cloudml-samples/molecules/data \
--outputs-dir /tmp/cloudml-samples/molecules/predictions
In alternativa, esegui la pipeline di previsione batch su Dataflow:
# For simplicity, we'll use the same files we used for training
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python predict.py \
--work-dir $WORK_DIR \
--model-dir $MODEL_DIR \
batch \
--project $PROJECT \
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--inputs-dir $WORK_DIR/data \
--outputs-dir $WORK_DIR/predictions
Quando la pipeline è in esecuzione, puoi visualizzarne l'avanzamento nell'interfaccia di monitoraggio di Dataflow:
Previsioni relative ai flussi di dati
Le previsioni di streaming sono ottimizzate per la latenza anziché per la velocità effettiva. Le previsioni di streaming funzionano meglio se fai previsioni sporadiche ma vuoi ottenere i risultati il prima possibile.
Il servizio di previsione (la pipeline di previsione di inserimento di flussi) riceve le molecole di input da un argomento Pub/Sub e pubblica l'output (previsioni) in un altro argomento Pub/Sub.
Crea l'argomento Pub/Sub di input:
gcloud pubsub topics create molecules-inputs
Crea l'argomento Pub/Sub di output:
gcloud pubsub topics create molecules-predictions
Esegui la pipeline di previsione di flussi di dati localmente:
# Run on terminal 1
PROJECT=$(gcloud config get-value project)
python predict.py \
--model-dir $MODEL_DIR \
stream \
--project $PROJECT
--inputs-topic molecules-inputs \
--outputs-topic molecules-predictions
In alternativa, esegui la pipeline di previsione per lo streaming su Dataflow:
# Run on terminal 1
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python predict.py \
--work-dir $WORK_DIR \
--model-dir $MODEL_DIR \
stream \
--project $PROJECT
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--inputs-topic molecules-inputs \
--outputs-topic molecules-predictions
Dopo aver avviato il servizio di previsione (la pipeline di previsione di streaming), devi eseguire un publisher per inviare le molecole al servizio di previsione e un sottoscrittore per ascoltare i risultati della previsione. L'esempio di codice Molecules fornisce servizi di publisher (publisher.py) e abbonati (subscriber.py).
L'editore analizza i file SDF da una directory e li pubblica nell'argomento di input. L'abbonato rimane in ascolto dei risultati della previsione e li registra. Per semplicità, in questo esempio vengono utilizzati gli stessi file SDF utilizzati nella fase di addestramento.
Esegui l'abbonato:
# Run on terminal 2
python subscriber.py \
--project $PROJECT \
--topic molecules-predictions
Esegui il publisher:
# Run on terminal 3
python publisher.py \
--project $PROJECT \
--topic molecules-inputs \
--inputs-dir $WORK_DIR/data
Dopo che l'editore inizia ad analizzare e pubblicare le molecole, inizierai a vedere le previsioni dell'abbonato.
Esegui la pulizia
Dopo aver eseguito la pipeline di previsioni di streaming, interrompi la pipeline per evitare addebiti.
Passaggi successivi
- Consulta la documentazione di Apache Beam
- Leggi la documentazione di TensorFlow:
- Prova un altro esempio di codice che utilizza Apache Beam e
tf.Transform. - Addestra un modello di machine learning per la classificazione delle serie temporali utilizzando l'esempio di Global Fishing Watch.