Nesta página, explicamos como executar um pipeline de análise genômica secundária no Google Cloud usando as práticas recomendadas do Genome Analysis Toolkit (GATK). As práticas recomendadas do GATK são fornecidas pelo Broad Institute (em inglês).
O fluxo de trabalho usado neste tutorial é uma implementação das práticas recomendadas do GATK referentes à descoberta de variantes nos dados de sequenciamento do genoma inteiro (WGS, na sigla em inglês). O fluxo de trabalho é gravado na linguagem de definição de fluxo de trabalho (WDL, na sigla em inglês) do Broad Institute e processado pelo executor da WDL, Cromwell (links em inglês).
Objetivos
Depois de concluir este tutorial, você saberá:
- Como executar um pipeline usando as práticas recomendadas do GATK com dados da versão 38 (em inglês) do genoma humano de referência
- Como executar um canal usando as práticas recomendadas do GATK com seus próprios dados
Custos
Neste documento, você usará os seguintes componentes faturáveis do Google Cloud:
- Compute Engine
- Cloud Storage
Para gerar uma estimativa de custo baseada na projeção de uso deste tutorial, use a calculadora de preços.
Antes de começar
- Faça login na sua conta do Google Cloud. Se você começou a usar o Google Cloud agora, crie uma conta para avaliar o desempenho de nossos produtos em situações reais. Clientes novos também recebem US$ 300 em créditos para executar, testar e implantar cargas de trabalho.
-
No console do Google Cloud, na página do seletor de projetos, selecione ou crie um projeto do Google Cloud.
-
Verifique se a cobrança está ativada para o seu projeto do Google Cloud.
-
Ative as APIs Cloud Life Sciences, Compute Engine, and Cloud Storage.
- Instale a CLI do Google Cloud.
-
Para inicializar a CLI gcloud, execute o seguinte comando:
gcloud init
- Atualize e instale os componentes
gcloud:gcloud components update
gcloud components install beta -
No console do Google Cloud, na página do seletor de projetos, selecione ou crie um projeto do Google Cloud.
-
Verifique se a cobrança está ativada para o seu projeto do Google Cloud.
-
Ative as APIs Cloud Life Sciences, Compute Engine, and Cloud Storage.
- Instale a CLI do Google Cloud.
-
Para inicializar a CLI gcloud, execute o seguinte comando:
gcloud init
- Atualize e instale os componentes
gcloud:gcloud components update
gcloud components install beta - Instale o git para fazer o download dos arquivos necessários.
-
Por padrão, o Compute Engine tem cotas de recursos para impedir o uso inadvertido. Ao aumentar as cotas, é possível iniciar
mais máquinas virtuais simultaneamente, o que aumenta a capacidade de processamento e reduz
o tempo de retorno.
Para os melhores resultados neste tutorial, você precisa solicitar mais cota acima do padrão do seu projeto. Veja recomendações para aumentos de cota na lista a seguir, além das cotas mínimas necessárias para executar o tutorial. Faça as solicitações de cota na região
us-central1:- CPUs: 101 (mínimo de 17)
- Padrão de disco permanente (GB): 10.500 (mínimo de 320)
- Endereços IP em uso: 51 (mínimo de 2)
É possível deixar outros campos de solicitação de cota vazios para manter suas cotas atuais.
Crie um bucket do Cloud Storage
Crie um bucket do Cloud Storage usando o comando gsutil mb. Devido a um requisito no mecanismo do Cromwell, não use um caractere de sublinhado (_) no nome do bucket ou você encontrará um erro.
gsutil mb gs://BUCKET
O pipeline gera resultados, registros e arquivos intermediários para esse bucket.
Fazer o download de arquivos de exemplo
Execute os seguintes comandos para fazer o download do WDL e do script auxiliar:
git clone https://github.com/broadinstitute/wdl-runner.git git clone https://github.com/gatk-workflows/broad-prod-wgs-germline-snps-indels.git
O repositório gatk-workflows/broad-prod-wgs-germline-snps-indels contém os seguintes arquivos necessários para executar o canal:
*.wdl: definição do fluxo de trabalho*.inputs.json: parâmetros de entrada, incluindo caminhos para os arquivos BAM e o genoma de referencial*.options.json: opções de ambiente de execução do fluxo de trabalho
O arquivo de definição de canal do Cromwell usado para executar canais WDL fica no repositório openwdl/wdl/runners/cromwell_on_google/.
Executar o canal usando dados de amostra
Nesta seção, mostramos como executar o pipeline com dados do WGS usando o build 38 do genoma de referência humana. Os arquivos de entrada são arquivos BAM desalinhados.
Para executar o pipeline, conclua as seguintes etapas:
Crie a variável de ambiente
GATK_GOOGLE_DIRque aponta para a pasta que contém os arquivos de canal Broad:export GATK_GOOGLE_DIR="${PWD}"/broad-prod-wgs-germline-snps-indelsCrie a variável de ambiente
GATK_OUTPUT_DIRque aponta para o bucket do Cloud Storage e uma pasta paraoutputdo fluxo de trabalho,logginge arquivos intermediárioswork:export GATK_OUTPUT_DIR=gs://BUCKET/FOLDER
Altere o diretório para a pasta
/wdl_runnerno repositório de que você fez o download. Esse diretório contém o arquivo de definição de pipeline para executar pipelines baseados em WDL no Google Cloud:cd wdl-runner/wdl_runner/
Execute o canal:
Escolha uma das seguintes opções dependendo se você está usando uma VPC padrão ou uma VPC personalizada:
VPC padrão
gcloud beta lifesciences pipelines run \ --pipeline-file wdl_pipeline.yaml \ --location us-central1 \ --regions us-central1 \ --inputs-from-file WDL=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.wdl,\ WORKFLOW_INPUTS=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.hg38.inputs.json,\ WORKFLOW_OPTIONS=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.options.json \ --env-vars WORKSPACE=${GATK_OUTPUT_DIR}/work,\ OUTPUTS=${GATK_OUTPUT_DIR}/output \ --logging ${GATK_OUTPUT_DIR}/logging/VPC personalizada
Crie as variáveis de ambiente
NETWORKeSUBNETWORKpara especificar o nome da sua rede VPC e da sub-rede:export NETWORK=VPC_NETWORK export SUBNETWORK=VPC_SUBNET
Edite o arquivo
PairedEndSingleSampleWf.options.jsonlocalizado no diretóriobroad-prod-wgs-germline-snps-indelse modifique as zonas para incluir apenas as que fazem parte da região da sub-rede. Por exemplo, se você estiver usando uma sub-redeus-central1, o campozonesserá"zones": "us-central1-a us-central1-b us-central1-c us-central1-f".gcloud beta lifesciences pipelines run \ --pipeline-file wdl_pipeline.yaml \ --location us-central1 \ --regions us-central1 \ --network ${NETWORK} \ --subnetwork ${SUBNETWORK} \ --inputs-from-file WDL=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.wdl,\ WORKFLOW_INPUTS=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.hg38.inputs.json,\ WORKFLOW_OPTIONS=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.options.json \ --env-vars WORKSPACE=${GATK_OUTPUT_DIR}/work,\ OUTPUTS=${GATK_OUTPUT_DIR}/output,\ NETWORK=${NETWORK},\ SUBNETWORK=${SUBNETWORK} \ --logging ${GATK_OUTPUT_DIR}/logging/
O comando retorna um ID de operação no formato
Running [operations/OPERATION_ID]. Use o comandogcloud beta lifesciences describepara rastrear o status do pipeline executando o seguinte comando (verifique se o valor da sinalização--locationcorresponde ao local especificado na etapa anterior):gcloud beta lifesciences operations describe OPERATION_ID \ --location=us-central1 \ --format='yaml(done, error, metadata.events)'O comando
operations describeretornadone: truequando o pipeline é concluído.É possível executar um script incluído com
wdl_runnerpara verificar a cada 300 segundos se o job está em execução, terminou ou retornou um erro:../monitoring_tools/monitor_wdl_pipeline.sh OPERATION_ID us-central1 300
Depois que o canal for concluído, execute o seguinte comando para listar as saídas no bucket do Cloud Storage:
gsutil ls gs://BUCKET/FOLDER/output/
Você pode ver os arquivos intermediários criados pelo canal e escolher os que quer manter, ou removê-los para reduzir os custos associados ao Cloud Storage. Para remover os arquivos, consulte Como excluir arquivos intermediários do bucket do Cloud Storage.
Executar o canal de práticas recomendadas do GATK nos dados
Antes de executar o canal nos dados locais, copie-os para um bucket do Cloud Storage.
Copiar arquivos de entrada
O canal pode ser executado com arquivos BAM desalinhados armazenados no Cloud Storage. Se os arquivos estiverem em um formato diferente, como FASTQ ou BAM alinhado, converta-os antes do upload para o Cloud Storage. É possível convertê-los localmente ou usar a API Pipelines (em inglês) para convertê-los na nuvem.
Veja no exemplo a seguir como copiar um único arquivo de um sistema de arquivos local para um bucket do Cloud Storage:
gsutil -m -o 'GSUtil:parallel_composite_upload_threshold=150M' cp FILE \
gs://BUCKET/FOLDER
Para mais exemplos de como copiar arquivos para um bucket do Cloud Storage, consulte a seção sobre Como copiar dados no Cloud Storage.
A ferramenta de linha de comando gsutil verifica checksums automaticamente. Desse modo, quando a
transferência for bem-sucedida, seus dados serão compatíveis para uso com as práticas
recomendadas do GATK.
Executar o canal nos dados
Para executar as práticas recomendadas do GATK nos arquivos BAM desalinhados, faça uma cópia de PairedEndSingleSampleWf.hg38.inputs.json (em inglês) e atualize os caminhos para apontar para os arquivos em um bucket do Cloud Storage.
Então, siga as etapas em Executar o pipeline com dados de amostra usando o arquivo PairedEndSingleSampleWf.hg38.inputs.json atualizado.
Se os dados não forem compostos de arquivos BAM desalinhados e contiverem genomas de referência, sequenciamento de exoma, painéis segmentados e dados somáticos, será preciso usar fluxos de trabalho diferentes. Consulte o Fórum de suporte do GATK e o repositório do GitHub do Broad Institute (links em inglês) para mais informações.
Solução de problemas
O canal está configurado para usar instâncias do Compute Engine em regiões e zonas específicas. Quando você executa a CLI gcloud, ela usa automaticamente uma região e zona padrão com base no local em que o projeto do Google Cloud foi criado. Isso poderá resultar na seguinte mensagem de erro quando você executar o canal:
"ERROR: (gcloud.beta.lifesciences.pipelines.run) INVALID_ARGUMENT: Error: validating pipeline: zones and regions cannot be specified together"Para resolver esse problema, remova a região e a zona padrão executando os comandos abaixo. Em seguida, execute o pipeline novamente:
gcloud config unset compute/zone gcloud config unset compute/regionPara mais informações sobre como definir a região e a zona padrão do seu projeto do Google Cloud, consulte Como alterar a região ou a zona padrão.
Se você encontrar problemas ao executar o pipeline, consulte a Solução de problemas da API do Cloud Life Sciences.
O GATK tem expectativas rigorosas sobre formatos de arquivo de entrada. Para evitar problemas, verifique se seus arquivos passaram pelo ValidateSamFile.
Se houver falha na execução do GATK, consulte os registros executando o seguinte comando:
gsutil ls gs://BUCKET/FOLDER/logging
Se você encontrar erros de permissão, verifique se a conta de serviço tem acesso de leitura para os arquivos de entrada e acesso de gravação para o caminho do bucket de saída. Se você estiver gravando arquivos de saída em um bucket em um projeto do Google Cloud que não é seu, precisará conceder permissão à conta de serviço para acessar o bucket.
Limpar
Como excluir arquivos intermediários do bucket do Cloud Storage
Quando você executa o pipeline, ele armazena arquivos intermediários em gs://BUCKET/FOLDER/work. É possível remover os arquivos após a conclusão do fluxo de trabalho para reduzir as cobranças do Cloud Storage.
Para visualizar a quantidade de espaço usado no diretório work, execute o comando
a seguir. A execução do comando pode levar vários minutos devido ao tamanho
dos arquivos no diretório.
gsutil du -sh gs://BUCKET/FOLDER/work
Para remover os arquivos intermediários do diretório work, execute o
seguinte comando:
gsutil -m rm gs://BUCKET/FOLDER/work/**
Excluir o projeto
A maneira mais fácil de evitar a cobrança é excluir o projeto usado no tutorial.
Para excluir o projeto, faça o seguinte:
- No Console do Google Cloud, acesse a página "Projetos".
-
Na lista de projetos, selecione o que você quer excluir e clique em Excluir projeto.
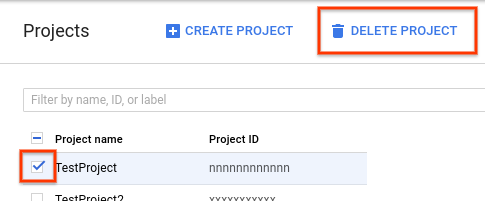
- Na caixa de diálogo, digite o código do projeto e clique em Encerrar para excluí-lo.
A seguir
- Neste tutorial, mostramos como executar um fluxo de trabalho predefinido em um caso de uso limitado, mas não deve ser executado na produção. Para mais informações sobre como executar o processamento de dados genômicos em um ambiente de produção no Google Cloud, consulte Arquitetura de referência de processamento de dados genômicos.
- O site do GATK e os fóruns do Broad Institute (links em inglês) incluem informações, documentação e suporte mais completos referentes às ferramentas do GATK e ao WDL.