Cette page explique comment exécuter un pipeline d'analyse génomique secondaire sur Google Cloud à l'aide des bonnes pratiques GATK (Genome Analysis Toolkit). Les bonnes pratiques GATK sont fournies par le Broad Institute.
Le workflow utilisé dans ce tutoriel est une mise en œuvre des bonnes pratiques GATK pour la découverte de variantes dans les données de séquençage du génome entier (WGS). Il est écrit dans le langage de définition de workflow (WDL) du Broad Institute et fonctionne sur l'exécuteur WDL Cromwell.
Objectifs
À la fin de ce tutoriel, vous saurez :
- exécuter un pipeline à l'aide des bonnes pratiques GATK avec les données issues de la version 38 du génome de référence humain ;
- exécuter un pipeline à l'aide des bonnes pratiques GATK avec vos propres données.
Coûts
Dans ce document, vous utilisez les composants facturables suivants de Google Cloud :
- Compute Engine
- Cloud Storage
Obtenez une estimation des coûts en fonction de votre utilisation prévue à l'aide du simulateur de coût.
Avant de commencer
- Connectez-vous à votre compte Google Cloud. Si vous débutez sur Google Cloud, créez un compte pour évaluer les performances de nos produits en conditions réelles. Les nouveaux clients bénéficient également de 300 $ de crédits gratuits pour exécuter, tester et déployer des charges de travail.
-
Dans Google Cloud Console, sur la page de sélection du projet, sélectionnez ou créez un projet Google Cloud.
-
Vérifiez que la facturation est activée pour votre projet Google Cloud.
-
Activer les API Cloud Life Sciences, Compute Engine, and Cloud Storage.
- Installez Google Cloud CLI.
-
Pour initialiser gcloudCLI, exécutez la commande suivante :
gcloud init
- Mettez à jour et installez les composants
gcloud:gcloud components update
gcloud components install beta -
Dans Google Cloud Console, sur la page de sélection du projet, sélectionnez ou créez un projet Google Cloud.
-
Vérifiez que la facturation est activée pour votre projet Google Cloud.
-
Activer les API Cloud Life Sciences, Compute Engine, and Cloud Storage.
- Installez Google Cloud CLI.
-
Pour initialiser gcloudCLI, exécutez la commande suivante :
gcloud init
- Mettez à jour et installez les composants
gcloud:gcloud components update
gcloud components install beta - Installez git pour télécharger les fichiers requis.
-
Par défaut, Compute Engine a mis en place des quotas de ressources qui permettent d'éviter toute utilisation accidentelle. En augmentant les quotas, vous pouvez lancer plus de machines virtuelles simultanément, ce qui augmente le débit et réduit les délais d'exécution.
Pour réussir au mieux ce tutoriel, vous devez demander un quota supplémentaire par rapport au quota par défaut de votre projet. La liste suivante contient des recommandations sur l’augmentation des quotas, ainsi que sur les quotas minimaux nécessaires à l’exécution du tutoriel. Effectuez vos demandes de quota dans la région
us-central1:- Processeurs : 101 (minimum 17)
- Disque persistant standard (Go) : 10 500 (minimum 320)
- Adresses IP en cours d'utilisation : 51 (minimum 2)
Vous pouvez laisser vides les autres champs de la demande pour conserver vos quotas actuels.
Créer un bucket Cloud Storage
Créez un bucket Cloud Storage à l'aide de la commande gsutil mb : N'utilisez pas de trait de soulignement (_) dans le nom du bucket. Vous risquez de rencontrer une erreur due à une exigence du moteur Cromwell.
gsutil mb gs://BUCKET
Le pipeline génère les résultats, les journaux et les fichiers intermédiaires dans ce bucket.
Télécharger les fichiers d'exemple
Exécutez les commandes suivantes pour télécharger le fichier WDL et le script d'aide :
git clone https://github.com/broadinstitute/wdl-runner.git git clone https://github.com/gatk-workflows/broad-prod-wgs-germline-snps-indels.git
Le dépôt gatk-workflows/broad-prod-wgs-germline-snps-indels contient les fichiers nécessaires à l'exécution du pipeline :
*.wdl: définition du workflow*.inputs.json: paramètres d'entrée, y compris les chemins d'accès aux fichiers BAM et au génome de référence*.options.json: options d'exécution du workflow
Le fichier de définition de pipeline Cromwell utilisé pour exécuter les pipelines WDL se trouve dans le dépôt broadinstitute/wdl-runner/wdl_runner/.
Exécuter le pipeline à l'aide de données d'échantillons
Cette section explique comment exécuter le pipeline avec des données WGS à l'aide de la version 38 du génome de référence humain. Les fichiers d'entrée sont des fichiers BAM non alignés.
Pour exécuter le pipeline, procédez comme suit :
Créez la variable d'environnement
GATK_GOOGLE_DIRqui pointe vers le dossier contenant les fichiers de pipeline Broad :export GATK_GOOGLE_DIR="${PWD}"/broad-prod-wgs-germline-snps-indelsCréez la variable d'environnement
GATK_OUTPUT_DIRqui pointe vers le bucket et un dossier Cloud Storage pour la sortie (output) du workflow, les fichiersworkintermédiaires et la journalisation (logging) :export GATK_OUTPUT_DIR=gs://BUCKET/FOLDER
Remplacez le répertoire par le dossier
/wdl_runnerdu dépôt que vous avez téléchargé. Ce répertoire contient le fichier de définition de pipeline pour l'exécution sur Google Cloud de pipelines basés sur WDL :cd wdl-runner/wdl_runner/
Exécutez le pipeline :
Choisissez l'une des options suivantes selon que vous utilisez un VPC par défaut ou un VPC personnalisé :
VPC par défaut
gcloud beta lifesciences pipelines run \ --pipeline-file wdl_pipeline.yaml \ --location us-central1 \ --regions us-central1 \ --inputs-from-file WDL=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.wdl,\ WORKFLOW_INPUTS=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.hg38.inputs.json,\ WORKFLOW_OPTIONS=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.options.json \ --env-vars WORKSPACE=${GATK_OUTPUT_DIR}/work,\ OUTPUTS=${GATK_OUTPUT_DIR}/output \ --logging ${GATK_OUTPUT_DIR}/logging/VPC personnalisé
Créez les variables d'environnement
NETWORKetSUBNETWORKpour spécifier les noms du réseau et du sous-réseau VPC :export NETWORK=VPC_NETWORK export SUBNETWORK=VPC_SUBNET
Modifiez le fichier
PairedEndSingleSampleWf.options.jsonsitué dans le répertoirebroad-prod-wgs-germline-snps-indels, puis modifiez les zones afin de n'inclure que les zones de la région de votre sous-réseau. Par exemple, si vous utilisez un sous-réseauus-central1, le champzonesdoit ressembler à ceci :"zones": "us-central1-a us-central1-b us-central1-c us-central1-f".gcloud beta lifesciences pipelines run \ --pipeline-file wdl_pipeline.yaml \ --location us-central1 \ --regions us-central1 \ --network ${NETWORK} \ --subnetwork ${SUBNETWORK} \ --inputs-from-file WDL=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.wdl,\ WORKFLOW_INPUTS=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.hg38.inputs.json,\ WORKFLOW_OPTIONS=${GATK_GOOGLE_DIR}/PairedEndSingleSampleWf.options.json \ --env-vars WORKSPACE=${GATK_OUTPUT_DIR}/work,\ OUTPUTS=${GATK_OUTPUT_DIR}/output,\ NETWORK=${NETWORK},\ SUBNETWORK=${SUBNETWORK} \ --logging ${GATK_OUTPUT_DIR}/logging/
La commande renvoie un ID d'opération au format
Running [operations/OPERATION_ID]. Vous pouvez utiliser la commandegcloud beta lifesciences describepour suivre l'état du pipeline. Exécutez pour cela la commande suivante (assurez-vous que la valeur de l'option--locationcorrespond à l'emplacement spécifié à l'étape précédente) :gcloud beta lifesciences operations describe OPERATION_ID \ --location=us-central1 \ --format='yaml(done, error, metadata.events)'La commande
operations describerenvoiedone: trueune fois le pipeline terminé.Vous pouvez exécuter un script fourni avec
wdl_runnerpour vérifier toutes les 300 secondes si la tâche est en cours d'exécution, si elle est terminée ou si elle a renvoyé une erreur :../monitoring_tools/monitor_wdl_pipeline.sh OPERATION_ID us-central1 300
Une fois le pipeline terminé, exécutez la commande suivante pour répertorier les résultats dans votre bucket Cloud Storage :
gsutil ls gs://BUCKET/FOLDER/output/
Vous pouvez afficher les fichiers intermédiaires créés par le pipeline et sélectionner ceux que vous souhaitez conserver, ou les supprimer pour réduire les coûts associés à Cloud Storage. Pour supprimer les fichiers, reportez-vous à la section Supprimer des fichiers intermédiaires du bucket Cloud Storage.
Exécuter le pipeline des bonnes pratiques GATK sur vos données
Avant d'exécuter le pipeline sur vos données locales, vous devez les copier dans un bucket Cloud Storage.
Copier les fichiers d'entrée
Le pipeline peut s'exécuter avec des fichiers BAM non alignés, stockés dans Cloud Storage. Si vous avez des fichiers dans un format différent, comme des fichiers BAM ou FASTQ alignés, vous devez les convertir avant de pouvoir les importer sur Cloud Storage. Vous pouvez réaliser cette opération localement ou utiliser l'API Pipelines pour les convertir dans le cloud.
L'exemple suivant montre comment copier un seul fichier d'un système de fichiers local vers un bucket Cloud Storage :
gsutil -m -o 'GSUtil:parallel_composite_upload_threshold=150M' cp FILE \
gs://BUCKET/FOLDER
Pour plus d'exemples sur la copie de fichiers dans un bucket Cloud Storage, consultez la section Copier des données dans Cloud Storage.
L'outil de ligne de commande gsutil vérifie les sommes de contrôle automatiquement. Ainsi, quand le transfert aboutit, vos données sont compatibles avec l'utilisation des bonnes pratiques GATK.
Exécuter le pipeline sur vos données
Pour exécuter les bonnes pratiques GATK sur vos propres fichiers BAM non alignés, effectuez une copie de PairedEndSingleSampleWf.hg38.inputs.json, puis une mise à jour des chemins d'accès à vos fichiers dans un bucket Cloud Storage.
Vous pouvez ensuite suivre les étapes présentées dans la section Exécuter le pipeline à l'aide de données d'échantillons en utilisant le fichier PairedEndSingleSampleWf.hg38.inputs.json.
Si vos données ne sont pas composées de fichiers BAM non alignés et contiennent des génomes de référence, un séquençage des exomes, des panels ciblés et des données somatiques, vous devez utiliser des workflows différents. Pour en savoir plus, consultez le forum d'assistance GATK et le dépôt GitHub du Broad Institute.
Dépannage
Le pipeline est configuré pour utiliser des instances Compute Engine dans des régions et des zones spécifiques. Lorsque vous exécutez gcloud CLI, elle utilise automatiquement une région et une zone par défaut en fonction de l'emplacement où votre projet Google Cloud a été créé. Il se peut que le message d'erreur suivant s'affiche lors de l'exécution du pipeline :
"ERROR: (gcloud.beta.lifesciences.pipelines.run) INVALID_ARGUMENT: Error: validating pipeline: zones and regions cannot be specified together"Pour résoudre ce problème, supprimez la région et la zone par défaut en exécutant les commandes suivantes, puis exécutez de nouveau le pipeline :
gcloud config unset compute/zone gcloud config unset compute/regionPour en savoir plus sur la configuration de la région et de la zone par défaut de votre projet Google Cloud, consultez la page Modifier la région ou la zone par défaut.
Si vous rencontrez des problèmes lors de l'exécution du pipeline, consultez la page Dépannage de l'API Cloud Life Sciences.
Les exigences de GATK sont strictes en matière de format des fichiers d'entrée. Pour éviter les problèmes, vous pouvez confirmer la validité de vos fichiers à l'aide de ValidateSamFile.
Si votre exécution GATK échoue, vous pouvez consulter les journaux à l'aide de la commande suivante :
gsutil ls gs://BUCKET/FOLDER/logging
Si vous rencontrez des erreurs d'autorisation, vérifiez que votre compte de service dispose de l'accès en lecture aux fichiers d'entrée et de l'accès en écriture au chemin d'accès du bucket de sortie. Si vous écrivez des fichiers de sortie dans un bucket situé dans un projet Google Cloud qui n'est pas le vôtre, vous devez autoriser le compte de service à accéder au bucket.
Nettoyer
Supprimer des fichiers intermédiaires du bucket Cloud Storage
Lorsque vous exécutez le pipeline, il stocke les fichiers intermédiaires dans le fichier gs://BUCKET/FOLDER/work. Vous pouvez supprimer les fichiers une fois le flux de travail terminé afin de réduire les frais générés par Cloud Storage.
Pour afficher la quantité d'espace utilisée dans le répertoire work, exécutez la commande suivante. L'exécution de la commande peut prendre plusieurs minutes en raison de la taille des fichiers du répertoire.
gsutil du -sh gs://BUCKET/FOLDER/work
Pour supprimer les fichiers intermédiaires du répertoire work, exécutez la commande suivante :
gsutil -m rm gs://BUCKET/FOLDER/work/**
Supprimer le projet
Le moyen le plus simple d'empêcher la facturation est de supprimer le projet que vous avez utilisé dans le cadre de ce tutoriel.
Pour supprimer le projet :
- Dans la console Google Cloud, accédez à la page "Projets".
-
Dans la liste des projets, sélectionnez celui que vous souhaitez supprimer, puis cliquez sur Delete project (Supprimer le projet).
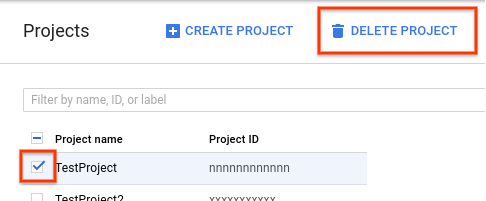
- Dans la boîte de dialogue, saisissez l'ID du projet, puis cliquez sur Arrêter pour supprimer le projet.
Étapes suivantes
- Ce tutoriel explique comment exécuter un workflow prédéfini dans un cas d'utilisation limité, mais n'est pas conçu pour être exécuté en production. Pour en savoir plus sur le traitement des données génomiques dans un environnement de production sur Google Cloud, consultez la page Architecture de référence du traitement des données génomiques.
- Le site GATK et les forums du Broad Institute fournissent des informations de contexte, une documentation et une assistance pour les outils GATK et WDL.