En esta página, se explica cómo ejecutar Sentieon® DNASeq® como una canalización de Google Cloud para el análisis genómico secundario. La canalización coincide con los siguientes resultados de la versión 3.7 de Genome Analysis Toolkit (GATK):
- Alineación
- Ordenamiento
- Eliminación de duplicados
- Recalibración del Nivel de calidad base (BQSR)
- Llamadas de variantes
Los formatos de entrada incluyen lo siguiente:
- archivos fastq
- Archivos BAM alineados y ordenados
Objetivos
Después de completar el instructivo, sabrás cómo realizar las siguientes actividades:
- Ejecutar una canalización en Google Cloud con Sentieon® DNAseq®
- Escribir archivos de configuración para distintos casos de uso de Sentieon® DNAseq®
Costos
En este documento, usarás los siguientes componentes facturables de Google Cloud:
- Compute Engine
- Cloud Storage
Para generar una estimación de costos en función del uso previsto, usa la calculadora de precios.
Antes de comenzar
- Instala Python 2.7 o superior. Para obtener más información sobre cómo configurar tu entorno de desarrollo de Python, incluida la instalación de pip en tu sistema, consulta la Guía de configuración del entorno de desarrollo de Python.
- Accede a tu cuenta de Google Cloud. Si eres nuevo en Google Cloud, crea una cuenta para evaluar el rendimiento de nuestros productos en situaciones reales. Los clientes nuevos también obtienen $300 en créditos gratuitos para ejecutar, probar y, además, implementar cargas de trabajo.
-
En la página del selector de proyectos de la consola de Google Cloud, selecciona o crea un proyecto de Google Cloud.
-
Asegúrate de que la facturación esté habilitada para tu proyecto de Google Cloud.
-
Habilita las API de Cloud Life Sciences, Compute Engine, and Cloud Storage.
-
En la página del selector de proyectos de la consola de Google Cloud, selecciona o crea un proyecto de Google Cloud.
-
Asegúrate de que la facturación esté habilitada para tu proyecto de Google Cloud.
-
Habilita las API de Cloud Life Sciences, Compute Engine, and Cloud Storage.
- Instala Google Cloud CLI.
-
Para inicializar la CLI de gcloud, ejecuta el siguiente comando:
gcloud init
- Instala y actualiza los componentes de
gcloud:gcloud components update
gcloud components install beta - Instala Git para descargar los archivos necesarios.
-
Con la configuración predeterminada, Compute Engine tiene cuotas de recursos para evitar el uso involuntario. Mediante el aumento de las cuotas, puedes iniciar más máquinas virtuales de forma simultánea, lo que aumenta la capacidad de procesamiento y reduce el tiempo de respuesta.
Para obtener los mejores resultados en este instructivo, debes solicitar una cuota adicional superior a la predeterminada de tu proyecto. En la siguiente lista, se proporcionan recomendaciones para los aumentos de cuota, así como las cuotas mínimas necesarias para ejecutar el instructivo. Realiza tus solicitudes de cuota en la región
us-central1:- CPU: 64
- Estándar de disco persistente (GB): 375
Puedes dejar vacíos otros campos de solicitud de cuota para mantener las cuotas actuales.
Licencia de evaluación de Sentieon®
Cuando usas esta canalización, Sentieon® te otorga automáticamente una licencia de evaluación gratuita de dos semanas que te permite utilizar su software con Google Cloud. Para recibir la licencia, ingresa tu dirección de correo electrónico en el campo EMAIL durante la configuración de la canalización. Consulta Comprende el formato de entrada para obtener información sobre cómo configurar este campo.
Si deseas seguir usando Sentieon® luego del vencimiento de la licencia de evaluación, comunícate con support@sentieon.com.
Instalar los requisitos previos y configura el entorno local
Si no tienes PendingIntent, ejecuta el siguiente comando para instalarlo con pip:
pip install virtualenv
Ejecuta el siguiente comando para crear un entorno aislado de Python e instalar dependencias:
virtualenv env source env/bin/activate pip install --upgrade \ pyyaml \ google-api-python-client \ google-auth \ google-cloud-storage \ google-auth-httplib2
Descarga la secuencia de comandos de la canalización
Ejecuta el siguiente comando para descargar los archivos de ejemplo y configurar su directorio actual:
git clone https://github.com/sentieon/sentieon-google-genomics.git cd sentieon-google-genomics
Comprende el formato de entrada
La canalización utiliza los parámetros especificados en un archivo JSON como su entrada.
En el repositorio que descargaste, hay un archivo examples/example.json con el siguiente contenido:
{
"FQ1": "gs://sentieon-test/pipeline_test/inputs/test1_1.fastq.gz",
"FQ2": "gs://sentieon-test/pipeline_test/inputs/test1_2.fastq.gz",
"REF": "gs://sentieon-test/pipeline_test/reference/hs37d5.fa",
"OUTPUT_BUCKET": "gs://BUCKET",
"ZONES": "us-central1-a,us-central1-b,us-central1-c,us-central1-f",
"PROJECT_ID": "PROJECT_ID"
"REQUESTER_PROJECT": "PROJECT_ID",
"EMAIL": "YOUR_EMAIL_HERE"
}
En la siguiente tabla, se describen las claves JSON del archivo:
| Clave JSON | Descripción |
|---|---|
FQ1 |
El primer par de lecturas en el archivo de entrada fastq. |
FQ2 |
El segundo par de lecturas en el archivo de entrada fastq. |
BAM |
El archivo de entrada BAM, si corresponde. |
REF |
El genoma de referencia. Si se los establece, se supone que los archivos de índice fastq/BAM existen. |
OUTPUT_BUCKET |
El bucket y el directorio que se usan para almacenar la salida de datos de la canalización. |
ZONES |
Una lista separada por comas de zonas de Google Cloud que se usarán para el nodo trabajador. |
PROJECT_ID |
Tu ID del proyecto de Google Cloud. |
REQUESTER_PROJECT |
Un proyecto para facturar cuando se transfieren datos de los buckets de pagos del solicitante |
EMAIL |
Tu dirección de correo electrónico |
Ejecuta la canalización
En el directorio
sentieon-google-genomics, edita el archivoexamples/example.jsony sustituye las variables BUCKET, REQUESTER_PROJECT, EMAIL y PROJECT_ID con los recursos correspondientes para tu proyecto de Google Cloud:{ "FQ1": "gs://sentieon-test/pipeline_test/inputs/test1_1.fastq.gz", "FQ2": "gs://sentieon-test/pipeline_test/inputs/test1_2.fastq.gz", "REF": "gs://sentieon-test/pipeline_test/reference/hs37d5.fa", "OUTPUT_BUCKET": "gs://BUCKET", "ZONES": "us-central1-a,us-central1-b,us-central1-c,us-central1-f", "PROJECT_ID": "PROJECT_ID", "REQUESTER_PROJECT": "PROJECT_ID", "EMAIL": "EMAIL_ADDRESS" }Configura la variable PROJECT_ID en tu entorno:
export PROJECT_ID=PROJECT_ID
Utiliza el siguiente comando para ejecutar la canalización DNAseq® en un conjunto de datos de prueba pequeño identificado mediante las entradas del archivo de configuración. Según la configuración predeterminada, la secuencia de comandos verifica que los archivos de entrada existen en el bucket de Cloud Storage antes de iniciar la canalización.
python runner/sentieon_runner.py --requester_project $PROJECT_ID examples/example.json
Si especificaste varios intentos interrumpibles, la canalización se reinicia cada vez que se interrumpe una instancia. Una vez que finaliza la canalización, se envía un mensaje a la consola que indica si la canalización se realizó correctamente o no.
Configuración recomendada
Para la mayoría de las situaciones, puedes optimizar el tiempo de respuesta y el costo con el uso de la siguiente configuración. La configuración ejecuta un genoma humano 30 veces a un costo de aproximadamente $1.25 y tarda alrededor de 2 horas. Un exoma humano completo cuesta alrededor de $0.35 y tarda alrededor de 45 minutos. Ambas estimaciones se basan en el hecho de que las instancias de la canalización no se van a interrumpir.
{
"FQ1": "gs://my-bucket/sample1_1.fastq.gz",
"FQ2": "gs://my-bucket/sample1_2.fastq.gz",
"REF": "gs://sentieon-test/pipeline_test/reference/hs37d5.fa",
"OUTPUT_BUCKET": "gs://BUCKET",
"BQSR_SITES": "gs://sentieon-test/pipeline_test/reference/Mills_and_1000G_gold_standard.indels.b37.vcf.gz,gs://sentieon-test/pipeline_test/reference/1000G_phase1.indels.b37.vcf.gz,gs://sentieon-test/pipeline_test/reference/dbsnp_138.b37.vcf.gz",
"DBSNP": "gs://sentieon-test/pipeline_test/reference/dbsnp_138.b37.vcf.gz",
"PREEMPTIBLE_TRIES": "2",
"NONPREEMPTIBLE_TRY": true,
"STREAM_INPUT": "True",
"ZONES": "us-central1-a,us-central1-b,us-central1-c,us-central1-f",
"PROJECT_ID": "PROJECT_ID",
"EMAIL": "EMAIL_ADDRESS"
}
Opciones adicionales
Puedes personalizar una canalización con el uso de las siguientes opciones.
Opciones de archivo de entrada
La canalización admite varios archivos fastq separados por comas como entrada, como se muestra en la siguiente configuración:
"FQ1": "gs://my-bucket/s1_prep1_1.fastq.gz,gs://my-bucket/s1_prep2_1.fastq.gz",
"FQ2": "gs://my-bucket/s1_prep1_2.fastq.gz,gs://my-bucket/s1_prep2_2.fastq.gz",
La canalización acepta archivos BAM separados por comas como entrada mediante la clave JSON BAM. Las lecturas en los archivos BAM no están alineadas con el genoma de referencia.
En cambio, comienzan en la etapa de anulación de duplicación de datos de la canalización. En el siguiente ejemplo, se muestra una configuración con dos archivos BAM como entrada:
"BAM": "gs://my-bucket/s1_prep1.bam,gs://my-bucket/s1_prep2.bam"
Configuración de datos del exoma completo o de conjuntos de datos grandes
Los ajustes en la configuración recomendada se optimizan para las muestran del genoma humano completo secuenciadas a una cobertura promedio de 30 veces superior. Para los archivos que son mucho más pequeños o más grandes que los conjuntos de datos estándares del genoma completo, puedes aumentar o disminuir los recursos disponibles para la instancia. Para obtener los mejores resultados con los conjuntos de datos grandes, utiliza la siguiente configuración:
{
"FQ1": "gs://sentieon-test/pipeline_test/inputs/test1_1.fastq.gz",
"FQ2": "gs://sentieon-test/pipeline_test/inputs/test1_2.fastq.gz",
"REF": "gs://sentieon-test/pipeline_test/reference/hs37d5.fa",
"OUTPUT_BUCKET": "gs://BUCKET",
"ZONES": "us-central1-a,us-central1-b,us-central1-c,us-central1-f",
"PROJECT_ID": "PROJECT_ID",
"EMAIL": "EMAIL_ADDRESS",
"DISK_SIZE": 600,
"MACHINE_TYPE": "n1-highcpu-64",
"CPU_PLATFORM": "Intel Broadwell"
}
En la siguiente tabla, se proporciona una descripción de la configuración utilizada:
| Clave JSON | Descripción |
|---|---|
DISK_SIZE |
Espacio SSD disponible para el nodo trabajador. |
MACHINE_TYPE |
El tipo de máquina virtual de Compute Engine que se usará. La configuración predeterminada es n1-standard-1. |
CPU_PLATFORM |
La plataforma de CPU que se debe solicitar. Debe ser un nombre de plataforma de CPU de Compute Engine válido (como "Intel Skylake"). |
Instancias interrumpibles
Puedes utilizar instancias interrumpibles en la canalización; para ello, configura la clave JSON PREEMPTIBLE_TRIES.
Según la configuración predeterminada, el ejecutor intenta ejecutar la canalización con una instancia estándar si se agotan los intentos interrumpibles o si la clave JSON NONPREEMPTIBLE_TRY se configura en 0. Para desactivar este comportamiento, establece la clave NONPREEMPTIBLE_TRY en false, como se muestra en la siguiente configuración:
"PREEMPTIBLE_TRIES": 2,
"NONPREEMPTIBLE_TRY": false
En la siguiente tabla se proporciona una descripción de la configuración utilizada:
| Clave JSON | Descripción |
|---|---|
PREEMPTIBLE_TRIES |
La cantidad de veces para intentar la canalización cuando se utilizan instancias interrumpibles. |
NONPREEMPTIBLE_TRY |
Determina si se intenta ejecutar la canalización con una instancia estándar después de que se agotan los intentos interrumpibles. |
Grupos de lectura
Los grupos de lectura se agregan cuando los archivos fastq están alineados con un genoma de referencia mediante Sentieon® BWA. Puedes proporcionar varios grupos de lectura separados por comas.
La cantidad de grupos de lectura debe coincidir con el número de archivos de entrada fastq.
El grupo de lectura predeterminado es @RG\\tID:read-group\\tSM:sample-name\\tPL:ILLUMINA.
Para cambiar el grupo de lectura, establece la clave READGROUP en el archivo de entrada JSON, como se muestra en la siguiente configuración:
"READGROUP": "@RG\\tID:my-rgid-1\\tSM:my-sm\\tPL:ILLUMINA,@RG\\tID:my-rgid-2\\tSM:my-sm\\tPL:ILLUMINA"
En la siguiente tabla, se proporciona una descripción de la configuración usada:
| Clave JSON | Descripción |
|---|---|
READGROUP |
Un grupo de lectura que contiene metadatos de muestra. |
Para obtener más información sobre los grupos de lectura, consulta Grupos de lectura.
Cómo transmitir archivos de entrada desde Cloud Storage
Puedes transmitir los archivos fastq de entrada desde Cloud Storage, lo que puede reducir el tiempo de ejecución total de la canalización. Para transmitir archivos fastq de entrada desde Cloud Storage, establece la clave JSON STREAM_INPUT en True:
"STREAM_INPUT": "True"
En la siguiente tabla, se proporciona una descripción de la configuración usada:
| Clave JSON | Descripción |
|---|---|
STREAM_INPUT |
Determina si se deben transmitir los archivos de entrada fastq directamente desde Cloud Storage. |
Marca de duplicado
Según la configuración predeterminada, la canalización quita las lecturas duplicadas de los archivos BAM. Para cambiar este comportamiento, configura la clave JSON DEDUP, como se muestra en la siguiente configuración:
"DEDUP": "markdup"
En la siguiente tabla, se proporciona una descripción de la configuración usada:
| Clave JSON | Descripción |
|---|---|
DEDUP |
Comportamiento de marcado de duplicaciones. Valores válidos:
|
Recalibración del nivel de calidad de las bases (BQSR) y sitios conocidos
La recalibración del nivel de calidad de las bases requiere sitios conocidos de variación genética. El comportamiento predeterminado es omitir esta etapa de la canalización. Sin embargo, puedes habilitar la BSQR si proporcionas sitios conocidos con la clave JSON BQSR_SITES. Si haces esto, puede usarse un archivo DBSNP para anotar las variantes de salida durante la llamada de variantes.
"BQSR_SITES": "gs://my-bucket/reference/Mills_and_1000G_gold_standard.indels.b37.vcf.gz,gs://my-bucket/reference/1000G_phase1.indels.b37.vcf.gz,gs://my-bucket/reference/dbsnp_138.b37.vcf.gz",
"DBSNP": "gs://sentieon-test/pipeline_test/reference/dbsnp_138.b37.vcf.gz"
En la siguiente tabla, se proporciona una descripción de la configuración utilizada:
| Clave JSON | Descripción |
|---|---|
BSQR_SITES |
Activa BSQR y utiliza una lista separada por comas de los archivos suministrados como sitios conocidos. |
DBSNP |
Se utiliza un archivo dbSNP durante la llamada de variantes. |
Intervalos
Para algunas aplicaciones, como la secuenciación orientada o de exoma completo, es posible que solo te interese una parte del genoma. En esos casos, proporcionar un archivo de intervalos objetivo puede acelerar el procesamiento y reducir las llamadas de variantes de baja calidad que no cumplen los requisitos. Puedes utilizar intervalos con las claves JSON INTERVAL_FILE y, además, INTERVAL.
"INTERVAL_FILE": "gs://my-bucket/capture-targets.bed",
"INTERVAL": "9:80331190-80646365"
En la siguiente tabla, se proporciona una descripción de la configuración utilizada:
| Clave JSON | Descripción |
|---|---|
INTERVAL_FILE |
Un intervalo que contiene intervalos genómicos para procesar. |
INTERVAL |
Una string que contiene un intervalo genómico para procesar. |
Opciones de salida
Según la configuración predeterminada, la canalización produce un archivo BAM procesado previamente, métricas de control de calidad y llamadas de variantes. Puedes inhabilitar cualquiera de estas salidas mediante las claves JSON NO_BAM_OUTPUT, NO_METRICS y NO_HAPLOTYPER. Si no se suministran los argumentos NO_HAPLOTYPER o NULL, puedes utilizar la clave JSON GVCF_OUTPUT para producir llamadas de variantes en formato gVCF en lugar de VCF.
"NO_BAM_OUTPUT": "true",
"NO_METRICS": "true",
"NO_HAPLOTYPER": "true",
"GVCF_OUTPUT": "true",
En la siguiente tabla, se proporciona una descripción de la configuración utilizada:
| Clave JSON | Descripción |
|---|---|
NO_BAM_OUTPUT |
Determina si se debe generar un archivo BAM procesado previamente. |
NO_METRICS |
Determina si se deben generar las métricas del archivo. |
NO_HAPLOTYPER |
Determina si se deben generar las llamadas de variantes. |
GVCF_OUTPUT |
Determina si se deben generar las llamadas de variantes en formato gVCF. |
Versiones de Sentieon® DNAseq
Puedes usar cualquier versión reciente del paquete de software de Sentieon® DNAseq® con la API de Cloud Life Sciences si especificas la clave JSON SENTIEON_VERSION, como en este ejemplo:
"SENTIEON_VERSION": "201808.08"
Las siguientes versiones son válidas:
201711.01201711.02201711.03201711.04201711.05201808201808.01201808.03201808.05201808.06201808.07201808.08
Limpia
Después de terminar el instructivo, puedes limpiar los recursos que creaste en Google Cloud para que no se te facturen en el futuro. En las siguientes secciones, se describe cómo borrarlos o desactivarlos.
Borra el proyecto
La manera más fácil de eliminar la facturación es borrar el proyecto que utilizaste para el instructivo.
Para borrar el proyecto, haz lo siguiente:
- En la consola de Google Cloud, ve a la página Proyectos.
-
En la lista de proyectos, selecciona el proyecto que quieres borrar y haz clic en Borrar proyecto.
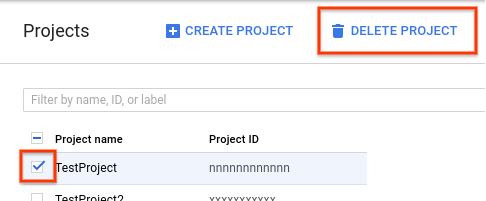
- En el cuadro de diálogo, escribe el ID del proyecto y, luego, haz clic en Cerrar para borrar el proyecto.
¿Qué sigue?
- Si tienes preguntas sobre la canalización o tienes algún problema, envía un correo electrónico a support@sentieon.com.