Neste tutorial, você verá como pré-processar, treinar e fazer previsões em um modelo de machine learning, usando o Apache Beam, o Google Dataflow e o TensorFlow.
Para demonstrar esses conceitos, usaremos a amostra de código do Molecules. Considerando dados moleculares como entrada, a amostra do código Molecules cria e treina um modelo de machine learning para prever a energia molecular.
Custos
Neste tutorial, há a possibilidade de usar componentes faturáveis do Google Cloud, incluindo um destes ou mais:
- Dataflow
- Cloud Storage
- AI Platform
Use a calculadora de preços para gerar uma estimativa de custo baseada na projeção de uso.
Visão geral
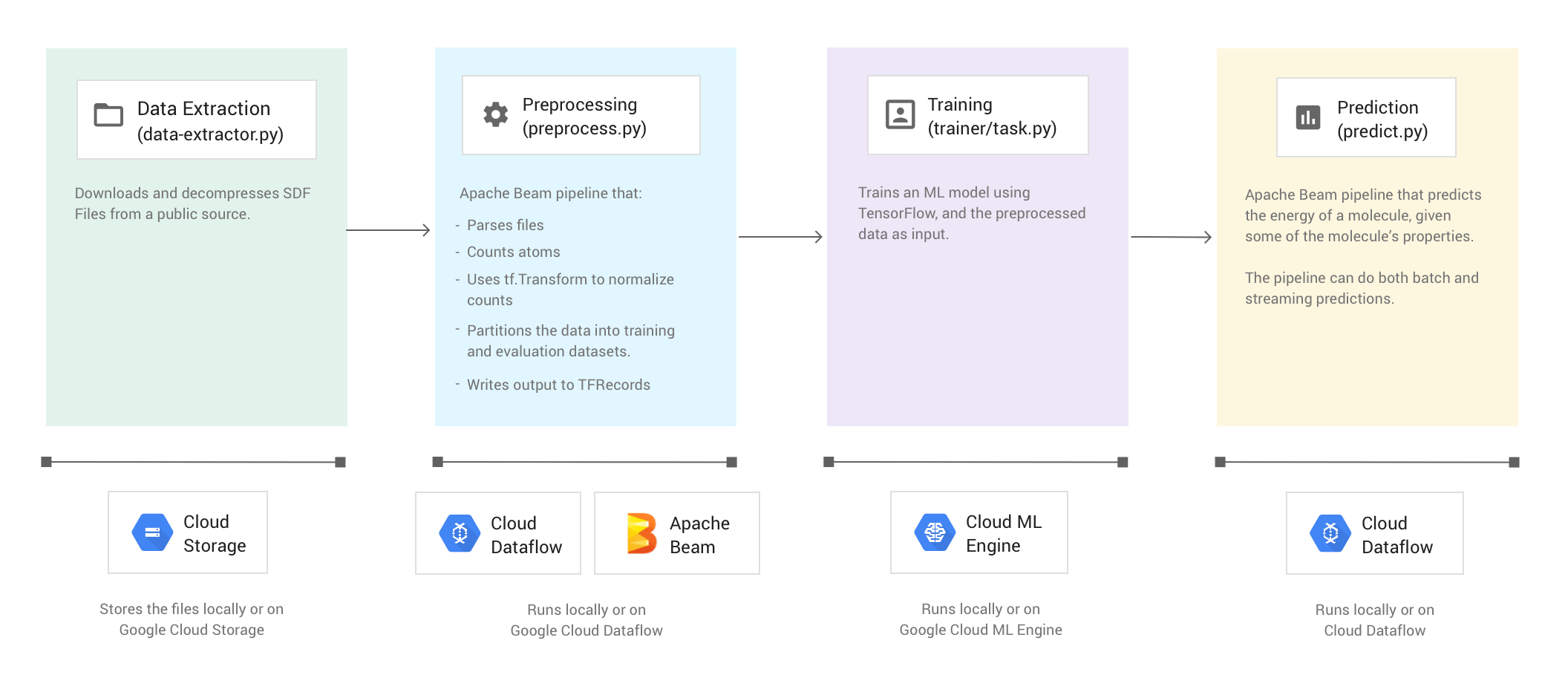
A amostra de código do Molecules extrai arquivos que contêm dados moleculares e conta o número de átomos de carbono, hidrogênio, oxigênio e nitrogênio em cada molécula. Em seguida, o código normaliza as contagens para valores entre 0 e 1 e alimenta os valores em um estimador de rede neural profunda do TensorFlow. O estimador de rede neural treina um modelo de aprendizado de máquina para prever energia molecular.
A amostra de código consiste em quatro fases:
- Extração de dados (
data-extractor.py) - Pré-processamento (
preprocess.py) - Treinamento (
trainer/task.py) - Previsão (
predict.py)
As seções abaixo analisam as quatro fases, mas este tutorial concentra-se mais nas fases que usam o Apache Beam e o Dataflow: a fase de pré-processamento e a de previsão. A fase de pré-processamento também usa a biblioteca de transformação do TensorFlow, conhecida como tf.Transform.
A imagem a abaixo mostra o fluxo de trabalho da amostra de código do Molecules.
Executar a amostra de código
Para configurar o ambiente, siga as instruções no README (em inglês) do repositório do GitHub do Molecules. Em seguida, execute a amostra de código do Molecules usando um dos scripts de wrapper fornecidos, run-local ou run-cloud. Esses
scripts executam automaticamente todas as quatro fases da amostra de código (extração, pré-processamento, treinamento e predição).
Como alternativa, você pode executar cada fase manualmente usando os comandos fornecidos nas seções deste documento.
Executar no local
Para executar a amostra de código do Molecules localmente, execute o script de wrapper run-local:
./run-local
Os registros de saída mostram quando o script executa cada uma das quatro fases (extração de dados, pré-processamento, treinamento e previsão).
O script data-extractor.py tem um argumento obrigatório para o número de arquivos.
Para facilitar o uso, os scripts run-local e run-cloud têm um padrão de cinco arquivos para esse argumento. Cada arquivo contém 25.000 moléculas. A execução da amostra de código leva aproximadamente de três a sete minutos do início ao fim. O tempo
de execução varia de acordo com a CPU do computador.
Executar no Google Cloud
Para executar a amostra de código do Molecules no Google Cloud, execute o script de wrapper
run-cloud. Todos os arquivos de entrada precisam estar no Cloud Storage.
Defina o parâmetro --work-dir para o bucket do Cloud Storage:
./run-cloud --work-dir gs://<your-bucket-name>/cloudml-samples/molecules
Fase 1: extração de dados
Código-fonte: data-extractor.py
O primeiro passo é extrair os dados de entrada. O arquivo data-extractor.py extrai e descompacta os arquivos SDF especificados. Em passos posteriores, o exemplo pré-processa esses arquivos e usa os dados para treinar e avaliar o modelo de machine learning. O arquivo extrai os arquivos SDF da fonte pública e os armazena em um subdiretório dentro do diretório de trabalho especificado. O diretório de trabalho padrão (--work-dir) é /tmp/cloudml-samples/molecules.
Armazenar os arquivos extraídos
Armazene os arquivos de dados extraídos no local:
python data-extractor.py --max-data-files 5
Ou armazene os arquivos de dados extraídos em um local do Cloud Storage:
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python data-extractor.py --work-dir $WORK_DIR --max-data-files 5
Fase 2: pré-processamento
Código-fonte: preprocess.py
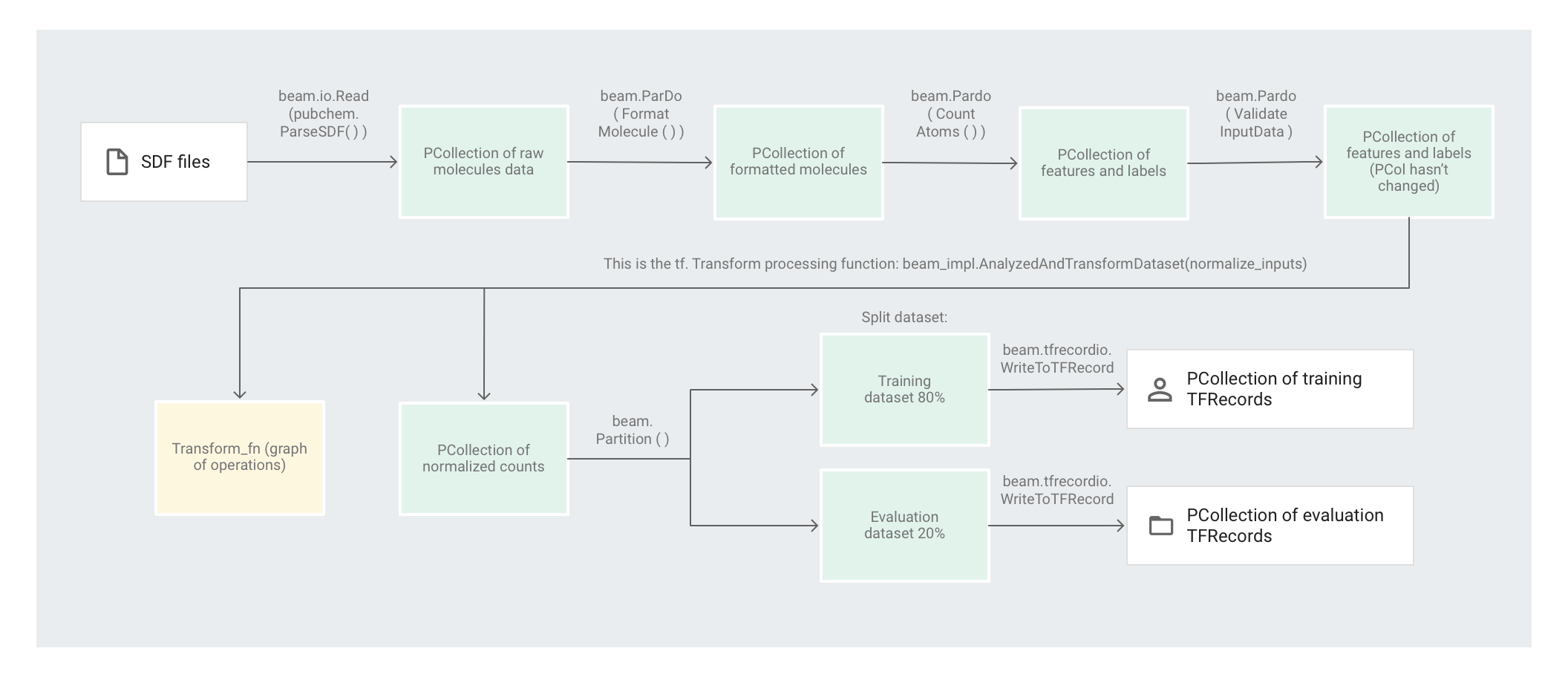
A amostra de código do Molecules usa um canal do Apache Beam para pré-processar os dados. O canal executa as ações de pré-processamento a seguir:
- Lê e analisa os arquivos SDF extraídos.
- Conta o número de átomos diferentes em cada uma das moléculas nos arquivos.
- Normaliza as contagens para valores entre 0 e 1 usando
tf.Transform. - Particiona o conjunto de dados em um conjunto de dados de treinamento e em um de avaliação.
- Grava os dois conjuntos de dados como objetos
TFRecord.
As transformações do Apache Beam podem manipular elementos individuais de uma só vez de maneira eficiente. No entanto, transformações que exigem uma passagem completa do conjunto de dados não são feitas facilmente apenas com o Apache Beam e têm melhores resultados com o uso do tf.Transform. Por causa disso, o código usa as transformações do Apache Beam para ler e formatar as moléculas, além de contar os átomos em cada molécula. Em seguida, o código usa tf.Transform para encontrar as contagens mínima e máxima globais e, assim, normalizar os dados.
A imagem a seguir mostra os passos no pipeline.
Como aplicar transformações baseadas em elementos
O código preprocess.py cria um pipeline do Apache Beam.
Em seguida, o código aplica uma transformação feature_extraction ao pipeline.
O pipeline usa SimpleFeatureExtraction como sua transformação feature_extraction.
A transformação SimpleFeatureExtraction, definida em pubchem/pipeline.py, contém uma série de transformações que manipulam todos os elementos de maneira independente.
Primeiro, o código analisa as moléculas do arquivo de origem, depois as formata para um dicionário de propriedades das moléculas e, por fim, conta os átomos nelas. Essas contagens são os atributos (entradas) para o modelo de machine learning.
A transformação de leitura beam.io.Read(pubchem.ParseSDF(data_files_pattern)) lê arquivos SDF de uma fonte personalizada (em inglês).
A fonte personalizada, chamada ParseSDF, é definida em pubchem/pipeline.py.
ParseSDF estende FileBasedSource (em inglês) e implementa o read_records que abre os arquivos SDF extraídos.
Ao executar a amostra de código do Molecules no Google Cloud, vários
workers (VMs)
podem ler os arquivos simultaneamente. Para garantir que não haja dois workers lendo o mesmo conteúdo nos arquivos, cada arquivo usa um range_tracker (em inglês).
O pipeline agrupa os dados brutos em seções de informações relevantes necessárias para os próximos passos. Cada seção no arquivo SDF analisado é armazenada em um dicionário (consulte pipeline/sdf.py), em que as chaves são os nomes das seções, e os valores são os conteúdos da linha não processada da seção correspondente.
O código aplica beam.ParDo(FormatMolecule()) ao pipeline. O ParDo aplica o DoFn chamado FormatMolecule a cada molécula. FormatMolecule produz um dicionário de moléculas formatadas. O snippet a seguir é um exemplo de um elemento na PCollection de saída:
{
'atoms': [
{
'atom_atom_mapping_number': 0,
'atom_stereo_parity': 0,
'atom_symbol': u'O',
'charge': 0,
'exact_change_flag': 0,
'h0_designator': 0,
'hydrogen_count': 0,
'inversion_retention': 0,
'mass_difference': 0,
'stereo_care_box': 0,
'valence': 0,
'x': -0.0782,
'y': -1.5651,
'z': 1.3894,
},
...
],
'bonds': [
{
'bond_stereo': 0,
'bond_topology': 0,
'bond_type': 1,
'first_atom_number': 1,
'reacting_center_status': 0,
'second_atom_number': 5,
},
...
],
'<PUBCHEM_COMPOUND_CID>': ['3\n'],
...
'<PUBCHEM_MMFF94_ENERGY>': ['19.4085\n'],
...
}
Em seguida, o código aplica beam.ParDo(CountAtoms()) ao pipeline. Os DoFnCountAtoms soma a quantidade de átomos de carbono, hidrogênio, nitrogênio e oxigênio que cada molécula tem. CountAtoms gera uma PCollection de atributos e rótulos.
Aqui está um exemplo de um elemento na PCollection de saída:
{
'ID': 3,
'TotalC': 7,
'TotalH': 8,
'TotalO': 4,
'TotalN': 0,
'Energy': 19.4085,
}
Então, o pipeline valida as entradas. O ValidateInputDataDoFn valida que cada elemento corresponda os metadados fornecidos ao input_schema. Essa validação
garante que os dados estejam no formato correto quando forem alimentados no TensorFlow.
Como aplicar transformações de passagem completa
A amostra de código do Molecules usa um Regressor de rede neural profunda para fazer predições. A recomendação geral é normalizar as entradas antes de alimentá-las no modelo de ML. O pipeline usa tf.Transform para normalizar as contagens de cada átomo para valores entre 0 e 1. Para ler mais sobre a normalização de entradas, consulte dimensionamento de atributos (em inglês).
A normalização dos valores requer uma passagem completa pelo conjunto de dados, gravando os valores mínimo e máximo. O código usa tf.Transform para percorrer todo o conjunto de dados e aplicar transformações de passagem completa.
Para usar tf.Transform, o código precisa fornecer uma função que contenha a lógica da transformação a ser executada no conjunto de dados. Em preprocess.py, o código usa a transformação AnalyzeAndTransformDataset fornecida por tf.Transform. Saiba mais sobre como usar tf.Transform.
Em preprocess.py, a função feature_scaling usada é normalize_inputs, que é definida em pubchem/pipeline.py. A função usa a função tf.Transform scale_to_0_1 para normalizar as contagens para valores entre 0 e 1.
É possível normalizar os dados manualmente, mas se o conjunto de dados for grande, será mais rápido usar o Dataflow. Usar o Dataflow permite que o pipeline seja executado em vários workers (VMs), conforme necessário.
Como particionar o conjunto de dados
Em seguida, o pipeline preprocess.py particiona o conjunto de dados único em dois. Ele aloca aproximadamente 80% dos dados a serem usados como dados de treinamento, e aproximadamente 20% deles como dados de avaliação.
Como gravar a saída
Por fim, o pipeline preprocess.py grava os dois conjuntos de dados (treinamento e avaliação) usando a transformação WriteToTFRecord (em inglês).
Executar o pipeline de pré-processamento
Execute o pipeline de pré-processamento localmente:
python preprocess.py
Ou execute o pipeline de pré-processamento no Dataflow:
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python preprocess.py \
--project $PROJECT \
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--work-dir $WORK_DIR
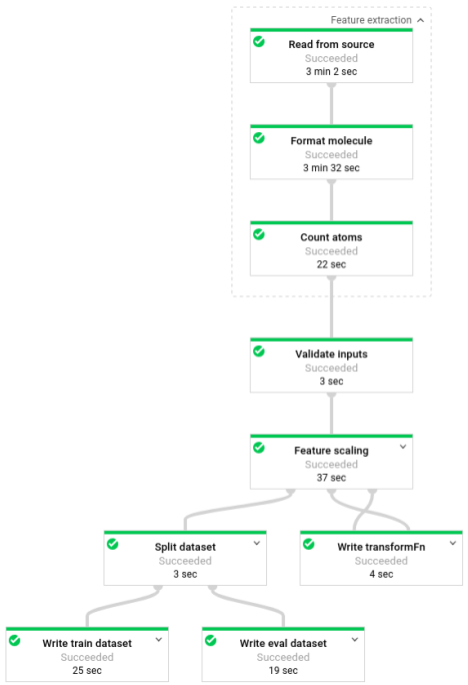
Depois que o pipeline estiver em execução, será possível visualizar o progresso dele na interface de monitoramento do Dataflow:
Fase 3: treinamento
Código-fonte: trainer/task.py
Lembre-se de que, no final da fase de pré-processamento, o código divide os dados em dois conjuntos de dados (treinamento e avaliação).
A amostra usa o TensorFlow para treinar o modelo de machine learning. O arquivo trainer/task.py na amostra de código de moléculas contém o código para treinar o modelo. A função principal de trainer/task.py carrega os dados que foram processados na fase de pré-processamento.
O Estimator usa o conjunto de dados de treinamento para treinar o modelo. Em seguida, usa o conjunto de dados de avaliação para verificar se o modelo prevê com precisão a energia molecular, dadas algumas das propriedades da molécula.
Saiba mais sobre como treinar um modelo ML.
Treinar o modelo
Treine o modelo localmente:
python trainer/task.py
# To get the path of the trained model
EXPORT_DIR=/tmp/cloudml-samples/molecules/model/export/final
MODEL_DIR=$(ls -d -1 $EXPORT_DIR/* | sort -r | head -n 1)
Ou treine o modelo no AI Platform:
- Primeiro, selecione uma região compatível para executar o job de treinamento
gcloud config set compute/region $REGION
- Iniciar o job de treinamento
JOB="cloudml_samples_molecules_$(date +%Y%m%d_%H%M%S)"
BUCKET=gs://<your bucket name>
WORK_DIR=$BUCKET/cloudml-samples/molecules
gcloud ai-platform jobs submit training $JOB \
--module-name trainer.task \
--package-path trainer \
--staging-bucket $BUCKET \
--runtime-version 1.13 \
--stream-logs \
-- \
--work-dir $WORK_DIR
# To get the path of the trained model:
EXPORT_DIR=$WORK_DIR/model/export
MODEL_DIR=$(gsutil ls -d $EXPORT_DIR/* | sort -r | head -n 1)
Fase 4: previsão
Código-fonte: predict.py
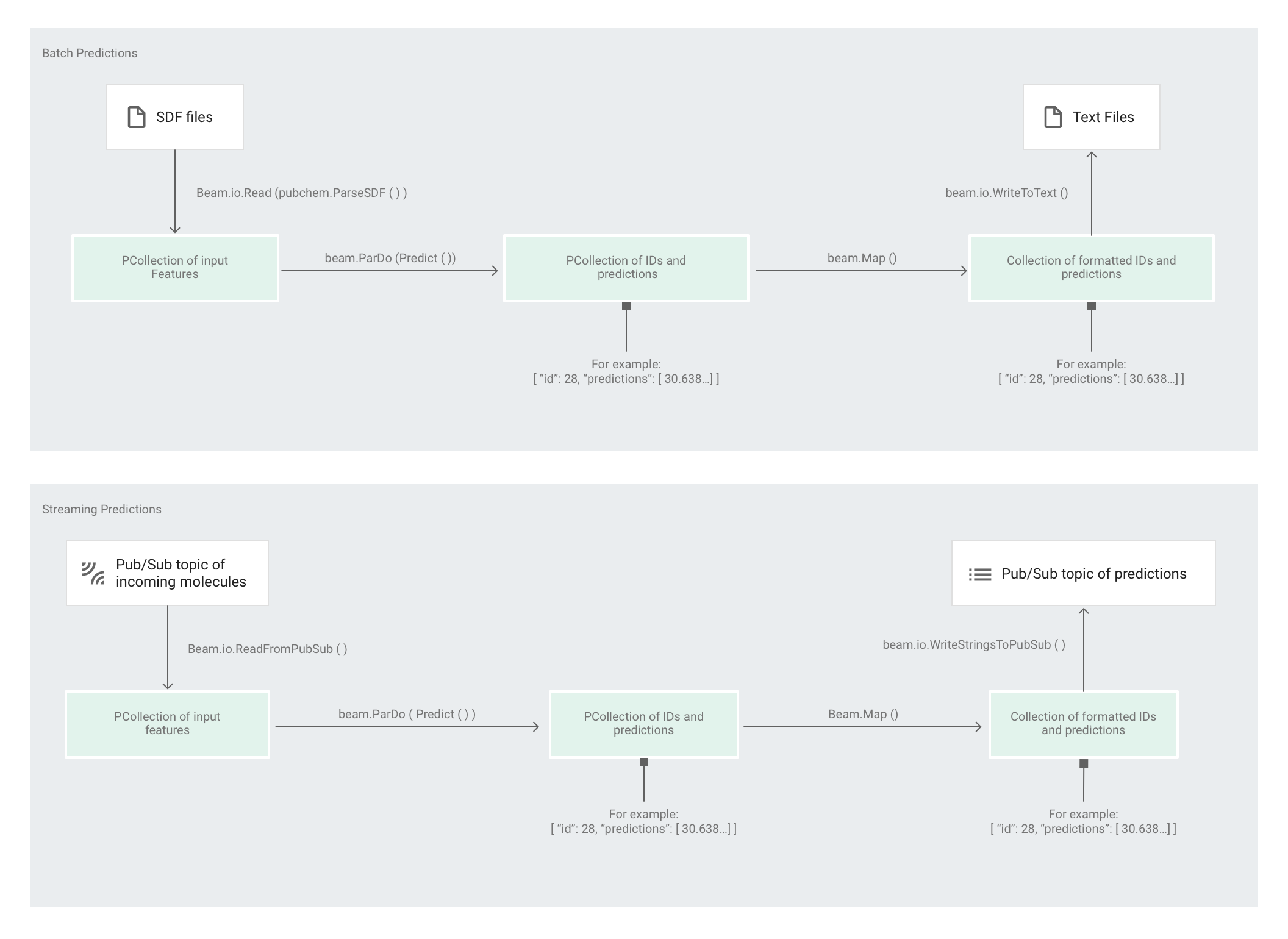
Depois que o Estimator treinar o modelo, forneça entradas para que ele faça previsões. Na amostra de código do Molecules, o pipeline em predict.py é responsável pelas previsões. O canal pode atuar como um canal em lote ou de streaming.
O código para o canal é o mesmo para lote e streaming, exceto para as interações de fonte e coletor. Se o pipeline for executado no modo em lote, ele lerá os arquivos de entrada da fonte personalizada e gravará as predições de saída como arquivos de texto no diretório de trabalho especificado. Se o pipeline for executado no modo de streaming, ele lerá as moléculas de entrada, conforme elas chegam, a partir de um tópico do Pub/Sub. O pipeline grava as previsões de saída, conforme elas ficam prontas, em um tópico diferente do Pub/Sub.
A imagem a seguir mostra as etapas nos canais de predição (lote e streaming).
Em predict.py, o código define o pipeline na função run:
O código chama a função run com os seguintes parâmetros:
Primeiro, o código passa a transformação pubchem.SimpleFeatureExtraction(source) como a transformação feature_extraction. Essa transformação, que também foi usada na fase de pré-processamento, é aplicada ao canal:
A transformação lê a fonte apropriada com base no modo de execução do pipeline (lote ou streaming), formata as moléculas e conta os átomos diferentes em cada molécula.
Em seguida, beam.ParDo(Predict(…)) é aplicado ao pipeline que realiza a previsão da energia molecular.
Predict, o DoFn que passou, usa o dicionário fornecido de atributos de entrada (contagem de átomos), para prever a energia molecular.
A próxima transformação aplicada ao pipeline é beam.Map(lambda result: json.dumps(result)), que pega o dicionário do resultado de previsão e o serializa em uma string JSON.
Por fim, a saída é gravada no coletor, como arquivos de texto no diretório de trabalho para o modo de lote ou como mensagens publicadas em um tópico do Pub/Sub para o modo de streaming.
Previsões em lote
As predições em lote são otimizadas por capacidade em vez de latência. Elas funcionam melhor se você estiver fazendo muitas predições e puder esperar que todas terminem antes de ter os resultados.
Executar o canal de predição no modo em lote
Execute o pipeline de previsão em lote localmente:
# For simplicity, we'll use the same files we used for training
python predict.py \
--model-dir $MODEL_DIR \
batch \
--inputs-dir /tmp/cloudml-samples/molecules/data \
--outputs-dir /tmp/cloudml-samples/molecules/predictions
Ou execute o pipeline de previsão em lote no Dataflow:
# For simplicity, we'll use the same files we used for training
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python predict.py \
--work-dir $WORK_DIR \
--model-dir $MODEL_DIR \
batch \
--project $PROJECT \
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--inputs-dir $WORK_DIR/data \
--outputs-dir $WORK_DIR/predictions
Depois que o pipeline estiver em execução, será possível visualizar o progresso dele na interface de monitoramento do Dataflow:
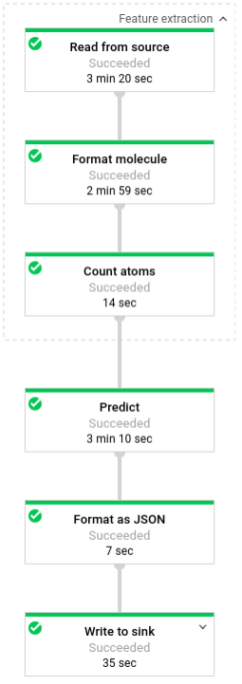
Previsões de streaming
As predições de streaming são otimizadas por latência em vez de capacidade. Elas funcionam melhor se você estiver fazendo previsões esporádicas, mas quiser os resultados o mais rápido possível.
O serviço de previsão (o pipeline de previsão de streaming) recebe as moléculas de entrada de um tópico do Pub/Sub e publica a saída (previsões) em outro tópico Pub/Sub.
Crie o tópico do Pub/Sub de entradas:
gcloud pubsub topics create molecules-inputs
Crie o tópico do Pub/Sub de saídas:
gcloud pubsub topics create molecules-predictions
Execute o pipeline de previsão de streaming localmente:
# Run on terminal 1
PROJECT=$(gcloud config get-value project)
python predict.py \
--model-dir $MODEL_DIR \
stream \
--project $PROJECT
--inputs-topic molecules-inputs \
--outputs-topic molecules-predictions
Ou execute o pipeline de previsão de streaming no Dataflow:
# Run on terminal 1
PROJECT=$(gcloud config get-value project)
WORK_DIR=gs://<your bucket name>/cloudml-samples/molecules
python predict.py \
--work-dir $WORK_DIR \
--model-dir $MODEL_DIR \
stream \
--project $PROJECT
--runner DataflowRunner \
--temp_location $WORK_DIR/beam-temp \
--setup_file ./setup.py \
--inputs-topic molecules-inputs \
--outputs-topic molecules-predictions
Depois de executar o serviço de previsão (o pipeline de previsão de streaming), é necessário executar um editor para enviar moléculas para o serviço de previsão e um assinante para escutar os resultados. O exemplo de código do Molecules fornece serviços de editor (publisher.py) e de assinante (subscriber.py).
O editor analisa arquivos SDF de um diretório e os publica no tópico de entradas. O assinante escuta os resultados da predição e os registra. Para simplificar, neste exemplo usaremos os mesmos arquivos SDF usados na fase de treinamento.
Execute o assinante:
# Run on terminal 2
python subscriber.py \
--project $PROJECT \
--topic molecules-predictions
Execute o editor:
# Run on terminal 3
python publisher.py \
--project $PROJECT \
--topic molecules-inputs \
--inputs-dir $WORK_DIR/data
Depois que o editor começar a analisar e publicar moléculas, você começará a ver as predições do assinante.
Limpar
Depois de concluir a execução do canal de predições de streaming, interrompa o canal para evitar cobranças.
Próximas etapas
- Consulte a documentação do Apache Beam.
- Leia a documentação do TensorFlow:
- Veja
outra amostra de código
que usa o Apache Beam e
tf.Transform. - Treine um modelo de ML para classificação de série temporal usando o exemplo do Global Fishing Watch.